Treatment effective on model mice with severe form of the disease
Credit: Kobe University
A multi-institutional research group has succeeded in developing an exon-skipping therapy using nucleic-acid therapeutics for Alport syndrome, an incurable kidney disease that can progress to renal failure. Disease model mice were found to be completely responsive to the treatment.
Members of the Department of Pediatrics at Kobe University Graduate School of Medicine, including Project Professor NOZU Kandai, Assistant Professor YAMAMURA Tomohiko, Former Professor MATSUO Masafumi, Professor IIJIMA Kazumoto, Associate Professor TAKAOKA Yutaka of the Division of Medical Informatics and Bioinformatics, Kumamoto University’s Professor KAI Hirofumi, and Team leader TAKASATO Minoru’s group at the RIKEN Center for Developmental Biology. The research was conducted in collaboration with Daiichi Sankyo Co. Ltd.
The results were published in the international scientific journal ‘Nature Communications’ on June 2, 2020.
Main Points
- Alport syndrome is a hereditary disorder characterized by nephritis, hearing loss and eye abnormalities (such as anterior lenticonus or cataracts). X-linked (*2) Alport syndrome is the most common form and men in particular exhibit severe symptoms.
- 90% of men with the disease develop end stage kidney disease (ESKD) by the age of 40, necessitating renal replacement therapies such as dialysis or kidney transplants. Furthermore, this disease shows very strong genotype-phenotype correlation and patients with truncating variants such as nonsense mutations (*3) in the causal gene COL4A5 develop ESKD in their early twenties.
- The research team developed a treatment method for Alport syndrome patients with severe mutation in the causal gene. The treatment involves using exon skipping therapy by ASO to change the causal genetic mutation into a milder form.
- This treatment method suppressed kidney failure in disease model mice and significantly prolonged their life expectancy. This means that the treatment is effective in delaying the progression to renal failure in these mice.
- There is currently no cure for Alport syndrome. It is hoped that these research achievements can contribute greatly towards the development of the world’s first specialized treatment method for this disease.
Research Background
Alport syndrome (AS) is the second most commonly occurring hereditary kidney disease after autosomal dominant polycystic kidney disease (ADPKD). There is one case of Alport syndrome in every 5,000 to 10,000 people. It is characterized by hearing loss, eye abnormalities and kidney disease, often progressing to end stage kidney disease (ESKD). Alport syndrome is divided into three groups according to how it is inherited; X-linked AS, autosomal recessive AS and autosomal dominant AS with approximately 80% of cases being X-linked Alport Syndrome (XLAS). Although male patients with XLAS are likely to have severe symptoms and around 90% of them experience end stage renal failure by the age of 40, there is still no specialized treatment for Alport syndrome. XLAS develops due to mutations in the COL4A5 gene that encodes the type IV collagen α5 chain and there is no expression of α5(IV) proteins in the kidneys of male patients with this form of the disease.
Male XLAS patients with severe mutations (such as nonsense mutations) in the COL4A5 gene develop renal failure over 15 years earlier than those with minor mutations (such as missense mutations). In other words, it is thought that if severe mutations could be changed to minor mutations, this would make the symptoms of the disease milder.
Research Methodology
The research team set about developing an exon-skipping therapy using nucleic acid therapeutics (antisense oligonucleotides/ASO (*4)) to change severe mutations into minor ones (Figure 1). ASO therapies have received increasing attention in recent years and they have been used to successfully treat intractable inherited diseases such as spinal muscular atrophy and Duchenne muscular dystrophy.
α5(IV) are the most important proteins for the formation of each kidney’s glomerular basement membrane. In XLAS, there is a mutation in the COL4A5 gene that encodes this protein, thus resulting in abnormal α5(IV) production. Severe mutations in COL4A5 (such as nonsense mutations) in particular can lead to ESKD development when the patient is in their early 20s.
However, previous research by Project Professor Nozu et al. has shown that renal failure was delayed by ten years when the number of base deletions was a multiple of 3 (in-frame mutation). This prompted the researchers to investigate the possibility of reducing symptoms using ASO-mediated exon-skipping therapy to change severe mutations to minor mutations (in-frame mutation).
The majority of exons (*6) that make up COL4A5, the casual gene for Alport syndrome, have a number of bases that are a multiple of 3. Therefore, if there is a nonsense mutation, it is possible to replace it with an in-frame (multiple of 3) mutation by deleting the mutated exons during splicing.
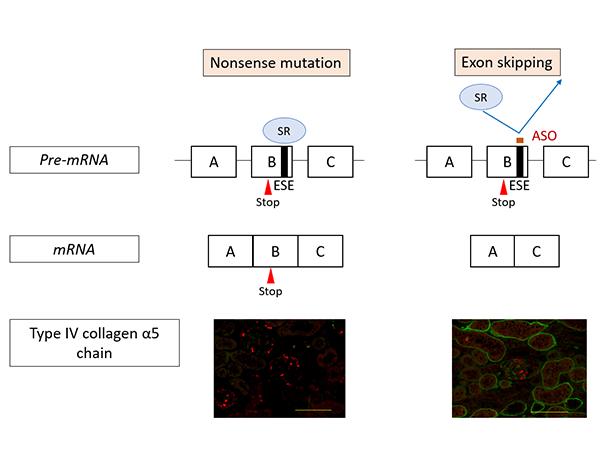
Figure 1 shows the action mechanism of exon-skipping by ASO. In nonsense mutation cases, the mutation remains in the messenger RNA (mRNA (*7)) resulting in the production of defective proteins. Consequently, α5(IV) proteins are not expressed in the kidneys (as shown on the left in Figure 1, the green signal that indicates the presence of α5(IV) proteins isn’t visible). On the other hand, under ASO-mediated exon-skipping therapy, ASO binds to the Exonic Splicing Enhancer (ESE) sites, blocking the binding of SR proteins (*8) to exons, at the pre-mRNA stage. SR protein binding is an important process for exon recognition and if the process is blocked that means that exon skipping can be induced. Consequently, mRNA was generated (bottom right of Figure 1) without a nonsense mutation. When this therapy was tested on model mice with nonsense mutations in COL4A5, α5(IV) protein expression was detected in the kidneys (Green signal in the bottom right of Figure 1).
The results for the ASO treated group and the vehicle treated group of model mice were compared. The mice receiving ASO treatment had lower urinary protein levels than the vehicle group and kidney failure (based on the level of serum creatinine) was suppressed (Figure 2). The researchers also succeeded in significantly prolonging life expectancy with this therapy (Figure 3). In addition to these results, microscopic examination of the kidney tissue indicated that they were able to significantly suppress the progression of renal damage.
The above results indicated that Alport syndrome model mice with severe genetic mutations were completely responsive to exon-skipping therapy.
Further Developments
Experiments using the animal model will be continued to determine the dosage and evaluate the drug’s safety. After this evaluation is completed, clinical trials on patients are scheduled to begin.
Acknowledgements
In December 2014, this study was selected by Daiichi Sankyo Co. Ltd.’s ‘TaNeDS’ program for cooperative research into drug discovery, and joint research and development with the company commenced. From April 2017 onwards, funding from the Japan Agency for Medical Research and Development (AMED) ‘Step 0’ (which supports exploratory research into revolutionary treatments and medical devices for intractable and rare diseases) enabled this study to be continued.
Glossary
*1: Nucleic-acid therapeutics: is a general term for medicines that are based on nucleotides (DNA and RNA components that control genetic information in living organisms). Nucleic-acid therapeutics have received great attention as a possible third generation treatment to follow on from low molecular weight heparin and antibody drugs. It is hoped that nucleic-acid therapeutics will support future medical treatment with their potential to cure diseases that have been difficult to treat with conventional medicine.
*2: X-linked form of a disease: A hereditary disease that occurs as a result of a mutation in a gene that coded the X chromosome. The symptoms are more severe in men because they only have one X chromosome, whereas women have two.
*3: Nonsense mutation: A nonsense mutation is where the substitution of a single base pair leads to the premature appearance of a stop codon. This causes the formation of proteins at the mutation site to be stopped halfway, resulting in the production of shortened and usually defective proteins. On the other hand, if a missense mutation occurs at the same mutation site, this causes a different amino acid to be incorporated into the protein, however the proteins are fully developed. This is why the most severe form of XLAS is caused by nonsense mutations.
*4: In-frame mutation: Every 3 bases encode one amino acid. If a deletion mutation occurs for a number of bases that are a multiple of 3, then the corresponding amino acids will be deleted but the resulting transcription will be normal. When the mutation after deletions is the same as a normal amino acid transcript, this is known as an in-frame mutation. XLAS patients with this kind of mutation have milder symptoms than those with nonsense mutations.
*5: Antisense nucleotide (ASO): In antisense oligonucleotide therapies, chemically engineered oligonucleotides that are complementary to specific messenger RNA are used to control gene expression. This allows gene sequences that are known to cause certain genetic disorders to be inactivated.
*6: Exon: Genes are often called ‘protein blueprints’ and are formed based on the sequence of particular bases. Exons are protein-coding transcripts and are thought to be the most important component of genes. The non-coding sections are called introns.
*7: Messenger RNA: Messenger RNA transcribes and splices the exon sequences from ‘protein blueprint’ genes. Messenger RNA forms proteins by incorporating each type of amino acid according to the base sequence.
*8: SR proteins: Pre-messenger RNA is produced from the transcription of part of the DNA and is made up of introns and exons. Subsequently, pre-messenger RNA is processed into messenger RNA via splicing. Splicing involves SR proteins binding to the exon areas and checking each exon. The introns are removed and messenger RNA is formed from the exons.
###
Media Contact
Verity Townsend
[email protected]
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}