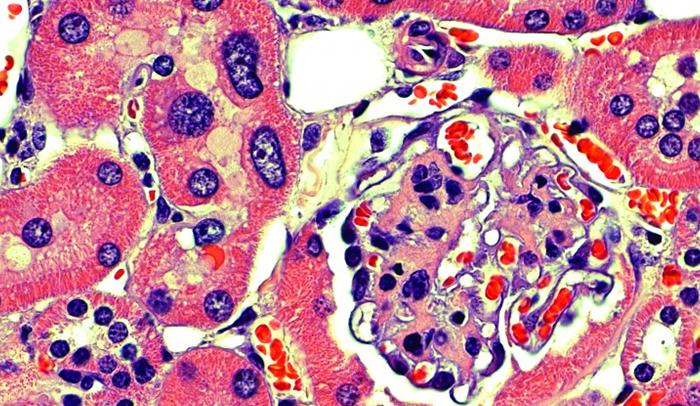
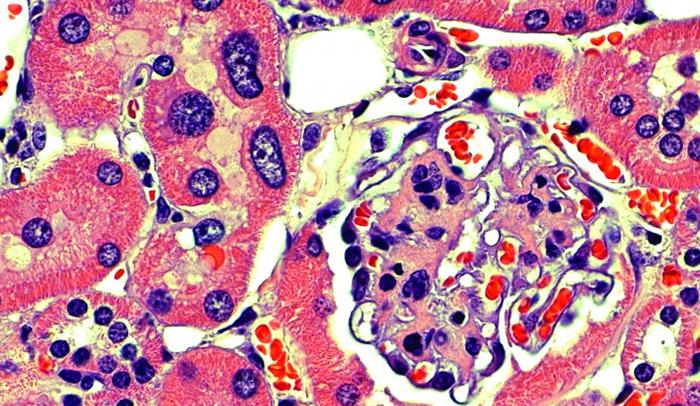
Like jewels in a vault, our precious genetic material is stored in the nucleus of a cell–sequestered away from potentially damaging cellular components and toxins so that no harm can come to it. Yet over the course of a life moving through this world, our DNA does get damaged, and our cells have a host of complicated repair mechanisms to deal with such injuries.
Agata Smogorzewska, head of the Laboratory of Genome Maintenance, wants to understand how cells repair interstrand cross-links, a particular type of DNA damage in which the two strands of the double helix that normally twine about each other become physically linked. This type of DNA damage is particularly problematic for the cell because when the two strands stick together, DNA cannot replicate and genes cannot be properly expressed.
In a recent study published in Genes and Development, Smogorzewska and her coworkers engineered mice that lack one of the genes involved in the repair process. These mice turned out to be particularly susceptible to drugs that induce DNA cross-links, because their cells don’t have a functional DNA repair system. Using these mice, the researchers will be able to further refine the detailed mechanisms involved in the repair of DNA damage.
Knockout mice as a disease model
Eliminating genes in mice is a standard technique for determining their function; the resultant animals are called knockout mice. The gene the researchers knocked out in the study is called FAN1, and a particularly interesting thing about it is the way in which it was initially implicated in DNA repair.
Mutations in many of the genes that regulate DNA interstrand cross-link repair cause Fanconi anemia, a rare disorder that can lead to infertility, bone marrow failure, and predisposition to cancer. To better understand what causes Fanconi anemia, Smogorzewska searched for genes involved in DNA interstrand cross-link repair. Unexpectedly, she found one that causes a completely different disease.
In humans, FAN1 deficiency causes karyomegalic interstitial nephritis, a disease that leads to kidney failure at around age 30. Thus far, the connection between the gene, which produces a DNA-cutting enzyme called a nuclease, and the kidney disease has remained obscure. The FAN1 knockout mice that Smogorzewska’s lab has generated allow them to investigate the role of FAN1 in DNA repair, and to figure out how mutations in the gene may lead to kidney disease.
In observing kidney cells from the FAN1 knockout mice, the researchers noticed that their nuclei were abnormally large. This phenomenon, called karyomegaly, has also been seen in patients with karyomegalic interstitial nephritis, and it’s thought to occur because the DNA duplicates too many times. The scientists saw these enlarged nuclei in the liver as well, and the mice developed liver dysfunction. They think the FAN1 protein helps protect liver function as the animals age.
“Both the kidney and the liver need to deal with a lot of toxins,” notes Smogorzewska, in ruminating about why FAN1 deficiency affects these tissues in particular. “FAN1 helps them do their job of detoxification. Without it, they can’t deal with the toxins.”
Independent DNA repair pathways
Even though FAN1 interacts with DNA repair proteins implicated in Fanconi anemia, this study demonstrates that it actually doesn’t have to do so in order to repair DNA damage. This may explain why karyomegalic interstitial nephritis and Fanconi anemia cause completely different symptoms in patients. Although FAN1 is clearly necessary for interstrand cross-link repair, the researchers say it is possible that the kidney and liver dysfunction are not directly related to a problem with this type of repair.
“FAN1 interacts with many different components in the cell nucleus and might be implicated in more genetic processes than what we previously thought,” Smogorzewska says. “That is why it is so important–and why it has been so hard–to really elucidate its function.”
Future directions
She notes that a lot remains to be learned about FAN1. “We don’t know what it actually does in the cell–what activates it, what it interacts with, and the specific type of DNA damage it repairs,” she says. “We don’t know what it is seeing that other nucleases aren’t seeing.” The mechanism of genome reduplication in the kidneys and livers is also opaque, and an area of intense study.
Now that they have a FAN1 knockout mouse in hand, and they have confirmed that it mimics many aspects of karyomegalic interstitial nephritis in humans, Smogorzewska’s lab can start to delve into how the loss of the gene causes kidney degeneration. For starters, the researchers would like to assess the nature of the toxins responsible for causing the DNA cross-links that patients with karyomegalic interstitial nephritis can’t handle.
Environmental toxins, including some herbal medicines known to cause kidney disease, don’t seem to be involved. So endogenous toxins–normal metabolic byproducts–are thought to be the most likely culprits. Smogorzewska plans to test whether FAN1 repairs DNA damaged by formaldehyde and acetaldehyde, an endogenous toxin known to induce DNA cross-links.
###
Media Contact
Katherine Fenz
[email protected]
212-327-7913
@rockefelleruniv
http://www.rockefeller.edu
The post Mice engineered with rare kidney disease shed light on how cells repair broken DNA appeared first on Scienmag.


{kind=link}