In the relentless battle against non-small cell lung cancer (NSCLC), targeted treatments such as epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) have ushered in a new era of hope, extending patient survival and improving quality of life. However, the initial promise of these therapies is frequently undermined by the development of drug resistance, a formidable clinical challenge that has stymied long-term treatment success. Recent groundbreaking research published in the British Journal of Cancer now illuminates a critical pathway behind this resistance, offering a beacon of hope for overcoming it.
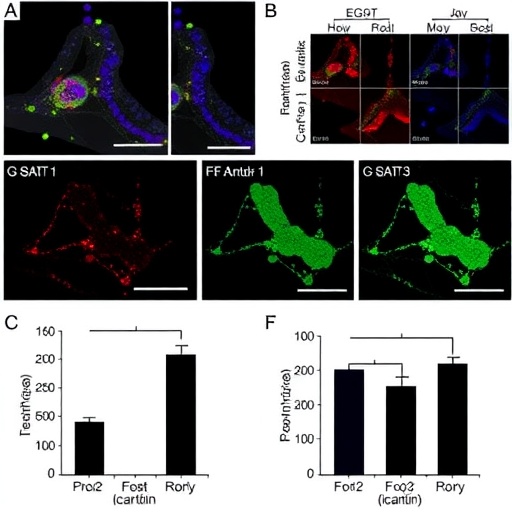
The study, spearheaded by Zhao, K., Zhang, J., Wang, R., and their colleagues, delves into the enigmatic role of Signal Transducer and Activator of Transcription 3 (STAT3) signaling in mediating resistance to EGFR-TKIs in NSCLC. STAT3, a transcription factor traditionally implicated in inflammation and cancer progression, emerges as a pivotal regulator of cellular behaviors linked to therapeutic failure. The researchers meticulously dissect the molecular interplay between STAT3 activation and the expression of stemness markers—biological indicators of a cell’s ability to self-renew and differentiate—as well as telomerase, the enzyme responsible for maintaining chromosomal integrity and promoting cellular immortality.
Drug resistance in NSCLC represents a multifaceted phenomenon where tumors evolve adaptive strategies to evade targeted therapies. EGFR-TKIs were initially celebrated for their precision in thwarting aberrant signaling in EGFR-mutated cancer cells, but over time these cells deploy compensatory pathways to survive. Activation of STAT3 signaling, as uncovered by Zhao et al., appears to serve as a master switch, orchestrating a suite of survival advantages. This mechanism involves upregulating genes associated with cancer stemness and telomerase activity, endowing the tumor cells with enhanced regenerative capacity and resistance to apoptotic signals induced by EGFR-TKI treatment.
In experimental models, the researchers observed that heightened STAT3 activity correlates strongly with increased expression of stem cell markers, including Sox2, Oct4, and Nanog—key players in maintaining the undifferentiated and highly plastic state of cancer cells. These markers not only confer therapeutic resilience but also contribute to tumor heterogeneity, a well-known culprit in drug resistance. Simultaneously, augmented telomerase activity ensures that tumor cells bypass replicative senescence, allowing for unchecked proliferation despite the presence of pharmacological inhibitors.
Perhaps the most groundbreaking aspect of this study lies in its exploration of icaritin, a natural compound derived from traditional Chinese medicine, which exhibits potent inhibitory effects on STAT3 signaling. Treatment with icaritin effectively reverses the stemness phenotype and downsizes telomerase expression, thereby restoring sensitivity to EGFR-TKIs in resistant NSCLC cells. This dual-targeted approach unravels a previously unappreciated therapeutic angle: disrupting the STAT3-mediated reinforcement of tumor cell immortality and plasticity to overcome drug resistance.
The implications of these findings are profound. The identification of STAT3 as a central mediator in EGFR-TKI resistance not only deepens the understanding of NSCLC biology but also opens the door for innovative combinational therapies. By integrating STAT3 inhibitors such as icaritin into existing treatment protocols, clinicians may be able to prevent or reverse resistance, thereby prolonging the effectiveness of EGFR-TKIs and enhancing patient outcomes. This strategy addresses the root of therapeutic failure rather than merely its symptoms, heralding a paradigm shift in lung cancer management.
Moreover, the study underscores the intricate crosstalk between signaling pathways and cellular phenotypes in cancer. The plasticity conferred by stemness markers enables tumor cells to adapt dynamically to environmental stressors, including drug treatment. Telomerase activation ensures these adaptive cells maintain their proliferative capacity over extended periods. Together, these features create a resilient cancer cellular ecosystem that conventional therapies struggle to dismantle.
What makes STAT3 particularly attractive as a therapeutic target is its widespread involvement in multiple pathways critical for tumor survival and progression. Unlike targeting a single mutation or downstream effector, inhibiting STAT3 can potentially disrupt the network of pro-survival signals, attenuating mechanisms beyond EGFR signaling alone. This multifaceted control may enhance the durability of therapeutic responses and mitigate the emergence of drug-resistant clones.
The translational potential of icaritin also merits attention. As a compound with established safety profiles in traditional medicine, its repurposing for lung cancer therapy could expedite clinical development and approval processes. The synergistic action of icaritin with EGFR-TKIs provides a compelling rationale for advancing to clinical trials, where patient stratification based on STAT3 activation status could refine personalized treatment plans.
This research also highlights the importance of integrating molecular diagnostics in cancer care. Detecting elevated STAT3 signaling or associated stemness markers could serve as a biomarker to identify patients at risk for developing resistance. Early intervention with STAT3 inhibitors might forestall resistance onset, improving prognoses and reducing the need for more aggressive, less targeted therapies.
Ultimately, the work of Zhao and colleagues bridges a critical gap between molecular oncology research and therapeutic innovation. Their elucidation of the STAT3-driven resistance mechanism equips the scientific community with a tangible target and a promising agent—icaritin—to counteract one of the most daunting hurdles in NSCLC treatment. As lung cancer remains a leading cause of cancer mortality worldwide, breakthroughs of this nature carry immense potential to save lives and transform clinical practice.
Future research building upon these findings is poised to explore the nuances of STAT3 regulation in diverse patient populations, potential resistance mechanisms against STAT3 inhibitors themselves, and the optimization of dosage regimens to maximize efficacy while minimizing toxicity. In addition, understanding how STAT3 interacts with other signaling cascades and the tumor microenvironment could reveal additional therapeutic vulnerabilities.
In an era where precision medicine strives to outpace cancer’s adaptability, targeting the fundamental drivers of therapy resistance represents a crucial frontier. The convergence of stemness, telomerase activity, and STAT3 signaling in NSCLC resistance presents a prime example of the complex biological challenges researchers confront. The promise of re-sensitizing tumors with compounds like icaritin emboldens the hope that drug resistance, once an insurmountable obstacle, may soon be rendered manageable through informed molecular interventions.
Through the rigorous experimental design and insightful analysis presented in this study, the scientific community gains a critical understanding of how lung cancer cells manipulate their internal circuitry to survive targeted therapies. Such knowledge not only advances the fight against NSCLC but also exemplifies the power of molecular biology to delineate and disrupt cancer’s defenses.
As clinical oncologists and researchers digest these findings, the path forward appears clear: integrated strategies that combine EGFR-TKIs with STAT3 pathway inhibitors hold the promise of transforming patient outcomes. The pursuit of such strategies will require collaboration across disciplines, from medicinal chemistry and molecular biology to clinical trial design and patient care.
In conclusion, the discovery that STAT3 signaling governs EGFR-TKI resistance through the regulation of stemness markers and telomerase, and that this resistance is reversible by icaritin, marks a milestone in lung cancer research. It invigorates the quest for durable, effective cancer therapies and exemplifies how understanding cancer’s molecular underpinnings can translate into tangible benefits for patients worldwide.
Subject of Research: The role of STAT3 signaling in mediating resistance to EGFR-tyrosine kinase inhibitors in non-small cell lung cancer through regulation of stemness markers and telomerase.
Article Title: STAT3 signaling mediates EGFR-TKI resistance in non-small cell lung cancer by regulating stemness markers and telomerase, reversed by icaritin.
Article References:
Zhao, K., Zhang, J., Wang, R. et al. STAT3 signaling mediates EGFR-TKI resistance in non-small cell lung cancer by regulating stemness markers and telomerase, reversed by icaritin. Br J Cancer (2026). https://doi.org/10.1038/s41416-026-03433-x
Image Credits: AI Generated
DOI: 10.1038/s41416-026-03433-x
Keywords: Non-small cell lung cancer, EGFR-tyrosine kinase inhibitors, STAT3 signaling, drug resistance, cancer stemness, telomerase, icaritin, targeted therapy, molecular oncology
Tags: cancer stem cell regulation in lung cancerEGFR TKI resistance mechanismsIcaritin cancer therapymolecular targets in lung cancer treatmentnon-small cell lung cancer drug resistancenovel treatments for EGFR-TKI resistant NSCLCovercoming EGFR inhibitor resistanceSTAT3 signaling pathway in lung cancerstemness markers in cancer cellstargeted therapy for NSCLCtelomerase role in cancer resistancetranscription factors in cancer progression

{kind=link}