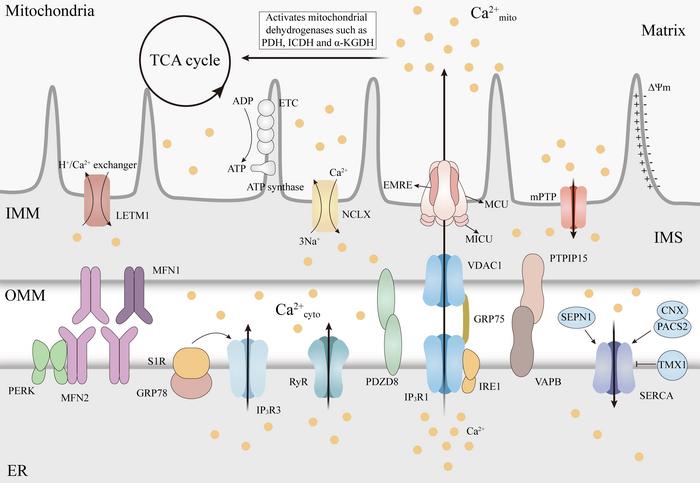

Mitochondria are recognized as pivotal organelles in maintaining cellular metabolism and signaling. Their role extends beyond merely producing ATP; they are central to regulating reactive oxygen species (ROS) generation and calcium (Ca²⁺) homeostasis. This intricate regulation of mitochondrial Ca²⁺ is essential for numerous cellular functions, yet its dysregulation can lead to severe pathological consequences, including neurodegenerative diseases. Understanding how mitochondrial Ca²⁺ influences the progression of such diseases could open new avenues for therapeutic interventions.
The interplay between mitochondrial Ca²⁺ uptake and efflux is a finely tuned process. The mitochondrial calcium uniporter (MCU) complex plays a crucial role in the influx of Ca²⁺ into mitochondria, allowing for metabolic activities and energy production. Conversely, the Na⁺/Ca²⁺ exchanger (NCLX) is responsible for Ca²⁺ efflux; thus, any disturbances in the activity of these mechanisms can result in mitochondrial Ca²⁺ overload. Furthermore, the communication between the endoplasmic reticulum (ER) and mitochondria through mitochondria-endoplasmic reticulum contact sites (MERCS) is vital for facilitating precise Ca²⁺ transfer. When this balance is disrupted, mitochondrial dysfunction may result, potentially leading to cell death.
Recent literature, particularly a review by researchers at the Chinese Academy of Sciences, underscores the implication of mitochondrial Ca²⁺ dysregulation in various neurodegenerative disorders. This includes well-studied pathologies such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and spinocerebellar ataxias (SCAs). These diseases display unique patterns of mitochondrial dysfunction, which are underpinned by the aberrant handling of Ca²⁺ within mitochondria.
In Alzheimer’s disease, for example, the aggregation of amyloid-beta (Aβ) proteins is known to disturb mitochondrial Ca²⁺ homeostasis. This disruption is characterized by increased Ca²⁺ uptake mediated by the MCU and a concomitant impairment of efflux through NCLX. As a consequence, the accumulation of ROS and energy depletion occur, ultimately leading to neuronal death. Moreover, alterations in MERCS serve to amplify this pathological cascade by enhancing the transfer of Ca²⁺ between the ER and mitochondria, pushing neuronal cells further toward apoptosis.
Parkinson’s disease provides another compelling example of mitochondrial dysfunction in neurodegeneration, where α-synuclein aggregates interfere with MERCS. This interference disrupts the normal Ca²⁺ transfer from the ER to mitochondria. The impact of genetic mutations in DJ-1, known for reducing antioxidant capacity, further compounds oxidative stress, posing additional challenges in maintaining mitochondrial health. These accumulated pathological processes highlight the critical role mitochondrial Ca²⁺ management plays within the disease context, making it a target for therapeutic strategies.
Huntington’s disease, driven by the mutant huntingtin (mHTT) protein resulting from CAG repeat expansions, similarly showcases the consequences of altered Ca²⁺ signaling. The heightened sensitivity of inositol trisphosphate receptor (IP₃R) and NMDA receptors induces abnormal Ca²⁺ signaling, which is implicated in mitochondrial dysfunction. The accumulating evidence suggests that it is not solely the presence of these genetic mutations but also how they disrupt ionic homeostasis that catalyzes disease progression.
Spinocerebellar ataxias, known for their hereditary nature caused by polyglutamine expansions, illuminate yet another facet of mitochondrial Ca²⁺ dysregulation. Mutant proteins exacerbate Ca²⁺ release from the ER through IP₃Rs, leading to excessive uptake by mitochondria and impaired efflux processes. The result is an aggregation of soluble toxic forms that can contribute to neuronal degeneration.
The mentioned review in the journal Mitochondrial Communications additionally raises the possibility of therapeutic interventions that target mitochondrial Ca²⁺ regulators. Promising strategies focus on the modulation of MCU and NCLX activities, stabilizing MERCS, or developing compounds that can prevent mitochondrial Ca²⁺ overload. These efforts include inhibitors of MCU and compounds aimed at stabilizing the mitochondrial permeability transition pore (mPTP). Although these approaches have shown promise in preclinical models, careful consideration of their specificity and impact on healthy tissues will be crucial in advancing to clinical applicability.
Importantly, while we emphasize the role of mitochondrial Ca²⁺ in pathophysiological contexts, it is equally essential to recognize its physiological significance. Author Tie-Shan Tang points out the challenge of developing pharmacological agents that selectively target the MCU complex, NCLX, or MERCS without affecting healthy cellular functions. This balancing act is a fundamental challenge within the biopharmaceutical landscape, underlining the need for comprehensive knowledge of mitochondrial dynamics.
As researchers continue to delineate the complexities surrounding mitochondrial Ca²⁺ in both healthy and diseased states, they uncover critical insights that could reshape therapeutic frameworks. The consensus remains clear: successfully targeting mitochondrial dysregulation has the potential to offer novel interventions that could change the course of neurodegenerative diseases, offering hope for effective management or even prevention.
Through international collaboration and continuously evolving methods in molecular biology and biochemistry, the field is poised to enhance our understanding of mitochondrial Ca²⁺ regulation. The goal remains clear: translate these complex scientific findings into effective treatments that nurture neuronal health and longevity while providing a deeper understanding of the underlying molecular mechanisms.
Drawing from this comprehensive review, it becomes evident how deeply intertwined mitochondrial health is with neurodegenerative processes. As our knowledge expands, we are reminded of the immense potential that lies within targeted interventions to combat these devastating conditions, fostering a future where neuroscience and cellular biology converge to foster health and well-being.
—
Subject of Research: Not applicable
Article Title: Decoding the influence of mitochondrial Ca2+ regulation on neurodegenerative disease progression
News Publication Date: Not specified
Web References: Not specified
References: Not specified
Image Credits: Sun et al.
Keywords: Mitochondria, Calcium Regulation, Neurodegenerative Diseases, Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease, Amyotrophic Lateral Sclerosis, Therapeutic Interventions, Cellular Biology, Molecular Biology, Health Science.
Tags: advancements in neurodegenerative disease researchcalcium homeostasis in neuronscalcium overload in mitochondriaendoplasmic reticulum and mitochondria communicationimplications of mitochondrial Ca²⁺ dysregulationmitochondrial calcium regulationmitochondrial calcium uniporter rolemitochondrial dysfunction and cell deathneurodegenerative disease mechanismsoxidative stress and neurodegenerationreactive oxygen species and neurodegenerationtherapeutic interventions for neurodegenerative diseases

{kind=link}