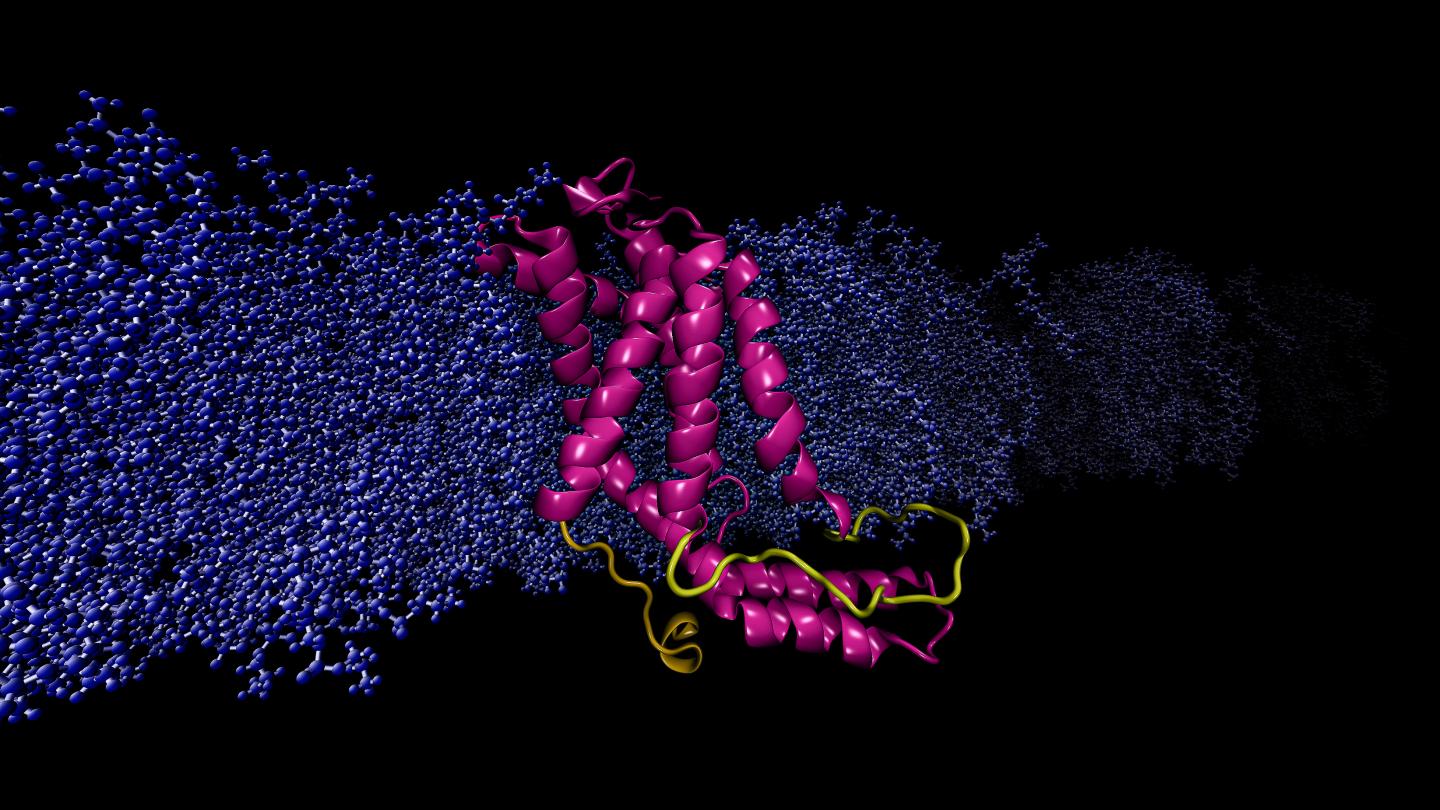
Researchers used a powerful supercomputer to run continuous, rapid computations simulating the molecular dynamics of a specific protein.
Credit: Image by Sogol Moradi
A new study by chemists at the University of Arkansas shows that X-ray crystallography, the standard method for determining the structure of proteins, can provide inaccurate information about a critical set of proteins – those found in cell membranes – which in turn could be leading to poor and inefficient drug design.
The researchers’ findings were published today in Scientific Reports, a Nature publication.
“Two-thirds of all drugs, including those used for chemotherapy, target proteins found on cell membranes,” said Mahmoud Moradi, assistant professor of chemistry and biochemstry in the J. William Fulbright College of Arts and Sciences. “Unfortunately, X-ray crystallography, the gold standard for determining the structure of proteins, has many limitations when dealing with those found in the cellular membrane. Our work exposes, and in many ways, explains these limitations.”
Considered the workhorse molecules of cells, proteins are responsible for nearly every task in living systems. Some proteins live inside cells, and some reside on the cell’s membrane, an outer layer of lipids that separates the cell from its external environment. Membrane proteins are critically important because they regulate the exchange of information and materials between the cell and its environment, a vital task for survival and normal function of the cell because any disorder in protein function can result in disease.
The study of protein function is necessary for understanding the molecular basis of disease. To do this, researchers have relied on X-ray crystallography, the primary tool for determining the shape and structure of proteins. X-ray crystallography is also essential for the purpose of designing drugs that efficiently manipulate the function of proteins. However, the study of membrane protein structure is difficult because their native environment is not compatible with X-ray crystallography. Researchers must remove the proteins from their native environment and place them in an artificial lipid environment before applying the technique.
Moradi and Thomas Harkey – an undergraduate student at the time and now a medical student at the University of Arkansas for Medical Sciences – addressed this problem from a different angle. For roughly two years, they used a supercomputer at the Arkansas High Performance Computing Center to run continuous, microsecond-level computations simulating the molecular dynamics of YidC2, a membrane protein with a crystallographically unresolved cytoplasmic loop in its molecular structure. Cytoplasmic loops are known to have functional significance in membrane proteins.
Moradi and Harkey’s simulations demonstrated that YidC2’s cytoplasmic loop stabilized the entire protein, particulary the C1 region, a potentially important area for drug design. Highly polar or charged lipid headgroups interacted with and stabilized the loop. This finding demonstrated that unresolved loops of membrane proteins could be important for the stabilization of proteins, despite the apparent lack of molecular structure.
“Typically, if part of a protein is not resolved in X-ray crystallography, it is interpreted as lacking a particular structure,” Moradi said. “We show that for membrane proteins and particularly parts of the protein that interact with the cell membrane, this interpretation is not accurate and could be misleading. We think that the alternative explanation for the disorder could be that the protein is not studied in its native membrane environment.”
Moradi said their results also demonstrated that computational chemistry and supercomputing technology can be used to model membrane proteins more accurately in an environment that mimics their physiological environment.
###
Media Contact
Mahmoud Moradi
[email protected]
479-575-6459
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}