
A groundbreaking study published in the April 2025 issue of Aging-US has unveiled critical insights into the molecular underpinnings of Werner syndrome (WS), a rare genetic disorder characterized by premature aging. The international research team, led by Sofie Lautrup and Evandro F. Fang from the University of Oslo and Akershus University Hospital, has discovered a direct link between deficient mitochondrial NAD+ levels and impaired cellular proliferation in WRN gene-deficient cells. This pioneering work not only advances our understanding of WS pathogenesis but also highlights the therapeutic potential of targeting NAD+ metabolism in age-related diseases.
Werner syndrome manifests clinically with characteristics typically observed in elderly individuals, including cataracts, osteoporosis, hair thinning, and cardiovascular disease, but with an onset as early as the third decade of life. The WRN gene, which encodes a helicase involved in DNA repair and genome maintenance, plays a protective role in cellular longevity. Its loss of function leads to accelerated cellular senescence and genomic instability. However, the exact mechanisms by which WRN deficiency drives premature aging at the mitochondrial and metabolic level have remained elusive—until now.
This new study reveals that cells lacking functional WRN protein suffer from a significant depletion of mitochondrial nicotinamide adenine dinucleotide (NAD+), a vital coenzyme central to energy metabolism, redox reactions, and mitochondrial health. NAD+ serves as a substrate for key enzymes involved in DNA repair, gene expression regulation, and metabolic adaptation. Deficiencies in mitochondrial NAD+ compromise oxidative phosphorylation, leading to reduced ATP production and enhanced mitochondrial dysfunction, which accelerates cellular aging features observed in WS.
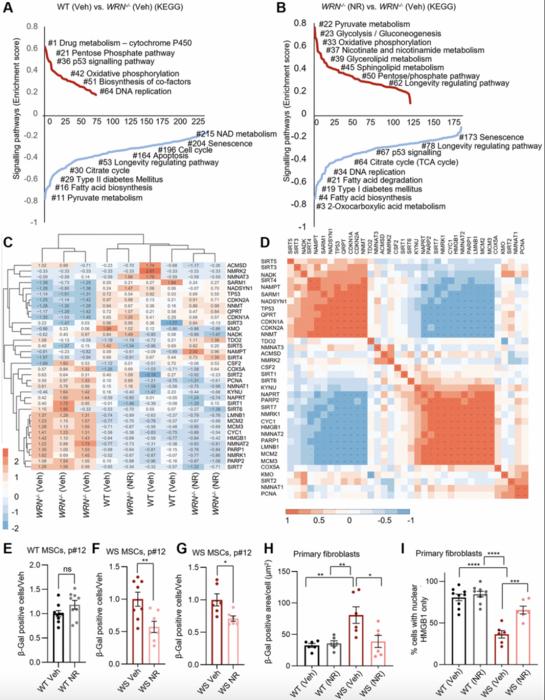
Through comprehensive gene-set enrichment analyses and transcriptomic profiling, the researchers identified that WRN-deficient mesenchymal stem cells (MSCs) exhibit widespread disruptions in metabolic and mitochondrial pathways. Notably, pathways governing NAD+ biosynthesis and salvage were significantly downregulated, suggesting that WRN plays a crucial role in maintaining intracellular NAD+ homeostasis. Intriguingly, treatment with nicotinamide riboside (NR), a precursor molecule that elevates cellular NAD+ levels, robustly rescued many of these metabolic defects within just 24 hours.
NR supplementation not only restored the expression of genes involved in mitochondrial function and proliferation but also mitigated cellular senescence markers in WS-derived MSCs and primary fibroblasts. Senescence-associated β-galactosidase (SA-β-Gal) staining, a gold-standard assay for detecting aging cells, showed a marked decrease in NR-treated WRN-deficient cells, confirming the rejuvenating effect of NAD+ augmentation. Additional assays demonstrated improved nuclear retention of HMGB1, a chromatin-associated protein whose cytoplasmic translocation is a hallmark of senescent cells, further corroborating the anti-senescence potential of NR.
Despite these promising results, the study carefully notes that NAD+ replenishment, while beneficial, did not completely reverse all dysfunctions in WRN-deficient cells. This finding underscores the multifaceted role of the WRN helicase, whose DNA repair and genome stability functions cannot be fully substituted by metabolic intervention alone. Nonetheless, the capacity of NR to partially restore cellular health highlights NAD+ metabolism as a viable therapeutic axis that could be exploited in mitigating premature aging syndromes.
Mechanistically, the interplay between WRN and NAD+ metabolism appears complex, involving coordinated regulation of genes that drive NAD+ biosynthetic pathways. Loss of WRN disrupts this balance, precipitating mitochondrial malfunctions and bioenergetic collapse that accelerate cellular aging. The findings also raise compelling questions about how subcellular NAD+ pools are regulated and distributed, and how these dynamics intersect with DNA repair and longevity pathways.
This work aligns with a growing body of research emphasizing the centrality of NAD+ homeostasis in aging and age-associated diseases such as neurodegeneration, metabolic disorders, and cancer. The ability to pharmacologically modulate NAD+ levels through precursors like NR or nicotinamide mononucleotide (NMN) has sparked considerable interest in developing novel anti-aging therapeutics. The present study strengthens this paradigm by providing concrete evidence that NAD+ augmentation can dampen senescence in the context of a defined genetic premature aging disorder.
Future investigations will be critical in unraveling the precise molecular crosstalk between WRN function, mitochondrial integrity, and NAD+ metabolism. Moreover, studies extending beyond in vitro models into animal systems and clinical settings will be invaluable to evaluate the translational potential of NAD+ boosting compounds for WS patients. If successful, such interventions could herald a new class of therapeutics aimed at mitigating cellular aging and extending healthspan in diverse human populations.
The implications of these discoveries extend far beyond Werner syndrome, offering valuable insight into the universal biological processes that regulate aging and cell vitality. By linking mitochondrial NAD+ depletion to proliferative defects and senescence, this research paves the way for targeted metabolic therapies that may one day combat the fundamental drivers of human aging.
In conclusion, Lautrup, Fang, and colleagues have provided compelling biological evidence that diminished mitochondrial NAD+ is a key contributor to premature cellular aging in WRN-deficient cells. Their work illuminates important pathways susceptible to intervention and offers hope for effective treatments that address the metabolic foundations of premature aging. This landmark study propels the field closer to harnessing metabolic modulation as a legitimate strategy in the fight against age-related decline and genetic aging disorders.
Subject of Research: Cells
Article Title: Decreased mitochondrial NAD+ in WRN deficient cells links to dysfunctional proliferation
News Publication Date: April 2, 2025
Web References: http://dx.doi.org/10.18632/aging.206236
Image Credits: Copyright © 2025 Lautrup et al., distributed under the Creative Commons Attribution License (CC BY 4.0)
Keywords: aging, Werner syndrome, premature aging, NAD+, mitochondria, proliferation
Tags: aging-related genetic disorderscellular senescence in genetic disordersimplications of WRN deficiencyinsights into mitochondrial health and longevityinternational research on agingmitochondrial function and agingNAD+ levels and cellular agingnicotinamide adenine dinucleotide metabolismpremature aging mechanisms in Werner syndrometherapeutic strategies for age-related diseasesWerner syndrome research findingsWRN gene and DNA repair


{kind=link}