Credit: TMIMS
Misfolding and aggregation of normally soluble proteins are common pathological features of many neurodegenerative diseases, including Alzheimer’s, Parkinson’s, Creutzfeldt-Jacob and Huntington’s diseases. For example, Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) are characterized by accumulation of misfolded α -synuclein in neuronal and/or glial cells, and therefore these diseases are termed α synucleinopathies. In PD and DLB, α-synuclein pathologies are mainly observed in neurons in the form of Lewy bodies and Lewy neurites, while glial cytoplasmic inclusions are seen in oligodendrocytes in MSA. Previous studies have suggested that α synuclein has the properties of prion, seed dependent aggregation, cell to cell propagation and the existence of distinct aggregate conformations (strains). There is a relationship between protein aggregate conformation and clinical phenotype in prion diseases, however, whether differences in the strains of α synuclein aggregates account for the different pathologies remained unclear. In this study, Suzuki and colleagues at TMIMS clarified a possible molecular mechanism to account for the different pathologies induced by different α synuclein strains.
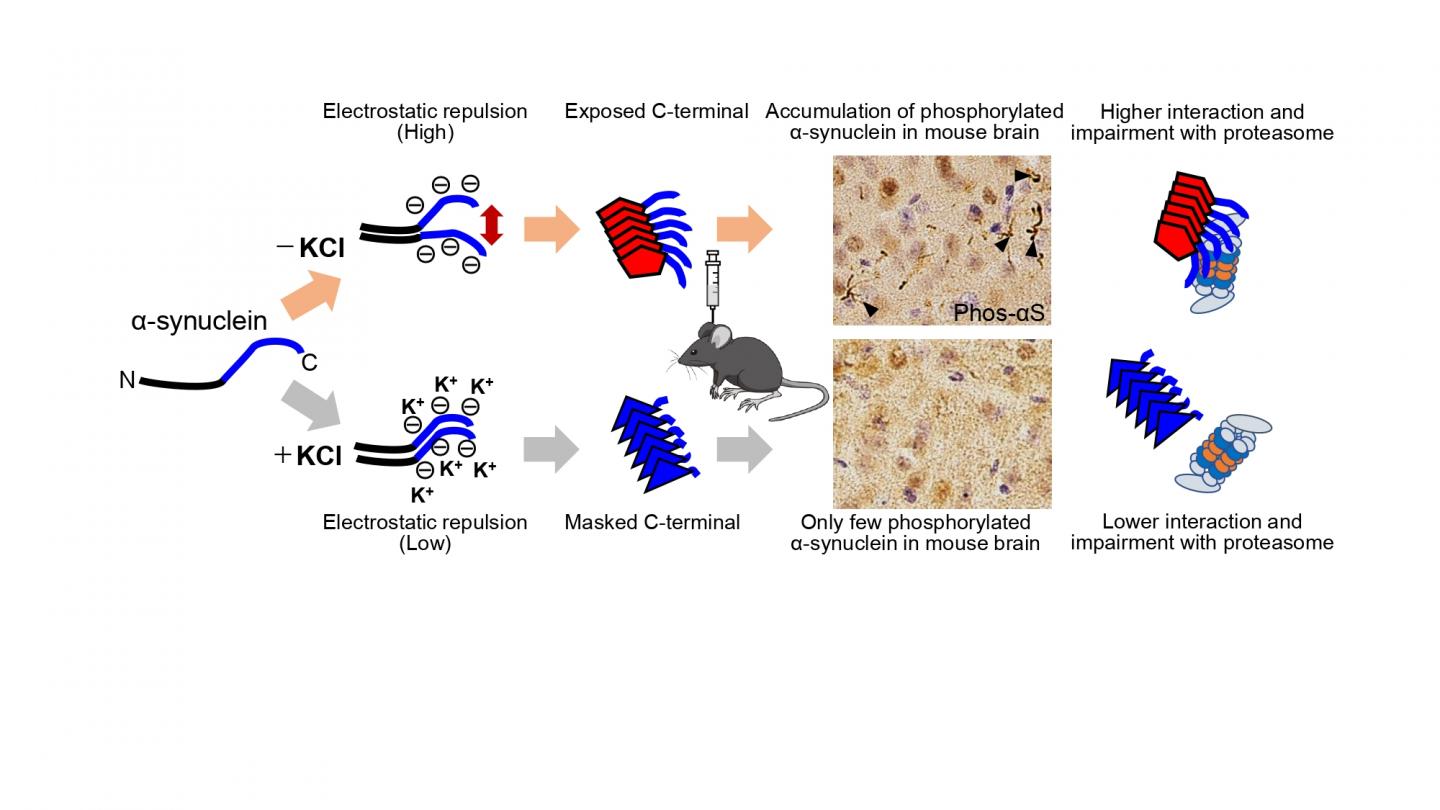
First, Suzuki and colleagues prepared recombinant α-synuclein monomer, agitated it in the presence or absence of salt at a physiological concentration and generated two types of α-synuclein fibrils, α-synuclein fibrils (+) and α-synuclein fibrils (-), from identical monomer. Next, they injected α-synuclein fibrils (+) and α-synuclein fibrils (-) into striatum of wild-type mice, and after one month, they examined the accumulation of phosphorylated α synuclein deposits resembling those observed in patients’ brains. They found that α-synuclein fibrils (-) induced Lewy body/Lewy neurite-like abnormal phosphorylated α-synuclein deposits through the mouse brain, whereas few phosphorylated α-synuclein deposits were induced by α-synuclein fibrils (+) (Figure). To further study the difference in the formation of pathological α-synuclein aggregates in neurons, Suzuki and colleagues compared the ability of the two α-synuclein strains to induce seed-dependent aggregation of α-synuclein in primary mouse cortical neurons. They observed a dramatic increase of phosphorylated α-synuclein accumulation, which is also positive for ubiquitin, only in the case of α-synuclein fibrils (-), while little accumulation was seen with α synuclein fibrils (+). They also found that not only ubiquitinated α-synuclein, but also other ubiquitinated proteins were accumulated in cells treated with α-synuclein fibrils (-), whereas there was no significant increase of ubiquitinated protein accumulation in cells treated with α-synuclein fibrils (+).
Next, Suzuki and colleagues examined proteasome activity in the presence of these two types of α-synuclein fibrils and found that only α-synuclein fibrils (-) apparently inhibited proteasome activity in vitro. They also investigated the interaction of 26S proteasome with the fibrils and found α-synuclein fibrils (-) co-precipitated with purified 26S proteasome complex. These results indicate that only α synuclein fibrils (-) interact with 26S proteasome and impair the proteasome activity.
Next, Suzuki and colleagues wanted to elucidate the structural difference of the two types of α-synuclein fibrils. They investigated the core regions and exposed regions of these α-synuclein fibrils and found that α-synuclein fibrils (-) have a smaller core region while α-synuclein fibrils (+) had a larger core region, extending to the C-terminal regions. This indicates that α-synuclein fibrils (-) have amyloid structure with a more exposed C-terminal region than α-synuclein fibrils (+). Thus, they considered that the C-terminal region of α-synuclein fibrils (-) might interact with 26S proteasome and impair its activities. To confirm this hypothesis, Suzuki and colleagues next examined the seeding activity and the effects on proteasome activity of C-terminally truncated α-synuclein fibrils formed in the absence of salt. They prepared C terminally 20-residue-truncated α-synuclein monomer (residues 1-120) and agitated it in the absence of salt. The resulting assemblies (αSΔC20 fibrils (-)) showed fibrillar morphology and then they treated primary mouse cortical neurons with αSΔC20 fibrils (-) and found only little accumulation of phosphorylated α-synuclein and ubiquitinated proteins. In addition, the catalytic activity of 26S proteasome was not impaired in the presence of αSΔC20 fibrils (-) and αSΔC20 fibrils (-) did not co-precipitated with 26S proteasome. Considering all these results, the C-terminal region of α-synuclein is exposed only in α synuclein fibrils (-) and this region interacts with 26S proteasome and impairs its activity.
The C-terminal region (residues 96-140) of α-synuclein is acidic and contains negatively charged residues, including aspartate and glutamate, as well as proline residues. When intermolecular repulsion at the C-terminal region is weakened by changes in ionic strength, exposure of the C-terminal region decreases, and more tightly packed α-synuclein fibrils are formed in the presence of salt. On the other hand, in the absence of salt, intermolecular repulsion at the C-terminal region causes the formation of α-synuclein fibrils in which the C-terminal region is exposed (Figure). Only this latter type of α-synuclein fibrils can interact with 26S proteasome complex in vitro, causing inhibition of 26S proteasome activity. If this also occurs in cells or brains, abnormal α synuclein aggregates, which might be partially degraded by proteasome, would be accumulated.
According to the prion hypothesis, differences in disease symptoms and lesions are caused by differences in the conformation of strains. Therefore, if α-synuclein is prion-like, differences in the structure of α-synuclein aggregates should cause the differences in the lesions observed in various α-synucleinopathies. In this study, Suzuki and colleagues confirmed that different pathologies were caused by two α-synuclein strains with different structures, and they also found that these α-synuclein strains differ in their ability to inhibit 26S proteasome activity. It is a noteworthy finding in this work that one of the two differently structured fibrils formed from identical monomer under different conditions inhibited proteasome, while the other did not. This clearly raises the possibility that inhibition of proteasome by abnormal α-synuclein plays a role in the pathology. These results provide a possible molecular mechanism to account for the different lesions induced by distinct α-synuclein strains. Considering that PD and MSA have different structures of accumulated α-synuclein, it seems highly likely that the structures of α-synuclein fibrils determine the lesions observed in these diseases.
###
Other institutions involved in the research are Tokyo Metropolitan University.
Media Contact
Genjiro Suzuki
[email protected]
Original Source
https:/
Related Journal Article
http://dx.



{kind=link}