Credit: Department of Neuropathology,
Medical Research Institute and Center for Brain Integration Research,
TMDU
Researchers led by Tokyo Medical and Dental University find that neuronal necrosis occurs much earlier in Alzheimer’s disease progression than originally thought, and uncover a novel target for future treatment strategies
Tokyo, Japan – Alzheimer’s remains the leading cause of dementia in Western societies, with some estimates suggesting that as many as 24 million people worldwide are living with the disease. Alzheimer’s is characterized by a progressive decline in cognitive ability that eventually affects even basic functions such as walking and swallowing. The exact cause of Alzheimer’s is unknown, but pathological changes in the brain, including neuron loss and an accumulation of protein aggregates called beta-amyloid plaques, are a diagnostic hallmark of Alzheimer’s disease.
Mild cognitive impairment (MCI) describes the slight but measurable changes in cognitive function that are often a precursor to Alzheimer’s disease. However, despite the importance of MCI, very little is known about the changes that occur in the brain during the progression from MCI to Alzheimer’s.
In a recent study published in Nature Communications, researchers led by Tokyo Medical and Dental University have now discovered that preventing pathological changes in the brain at the MCI stage could eliminate Alzheimer’s disease altogether.
“Neuronal death, is obviously very important in the development of Alzheimer’s, but is notoriously difficult to detect in real time because dying cells cannot be stained using chemical or immunohistological methods,” says lead author of the study Hikari Tanaka. “Because of this, we used a new biomarker called pSer46-MARCKS to detect degenerative neurites surrounding dying neurons, allowing us to quantify levels of necrosis, a prototype of neuronal death, at different stages of disease.”
Surprisingly, the researchers found that neuronal death occurred much earlier than originally thought, with higher levels of necrosis seen in patients with MCI than in patients with full-blown Alzheimer’s disease.
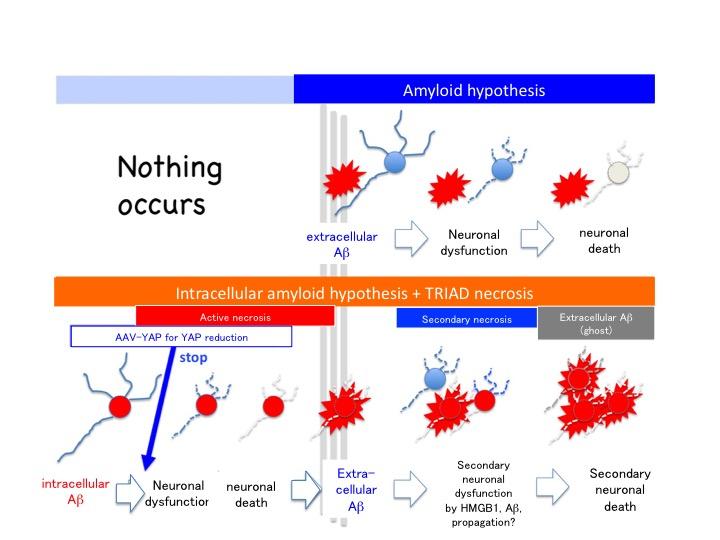
The researchers also observed a significant decrease in the levels of a protein known as YAP in Alzheimer’s disease model mice and human patients with MCI. YAP positively affects the activity of a second protein called TEAD, a deficiency of which leads to neuronal necrosis. Microscopic examination revealed that the missing YAP was sequestered within beta-amyloid plaques, which have also been linked to neuronal toxicity.
By directly injecting a gene therapy vector expressing YAP analog into the cerebral spinal fluid of mice that were genetically engineered to provide a model of Alzheimer’s, the researchers were able to prevent early-stage neuron loss, restore cognitive function, and prevent the development of beta-amyloid plaques.
“Confirming that neuronal necrosis was dependent on YAP was really the pivotal moment for us, but observing the almost transformative effects of YAP supplementation was hugely exciting,” says senior author of the study Hitoshi Okazawa. “By showing that neuronal necrosis is YAP-dependent and begins prior to the onset of most symptoms, we predict that novel Alzheimer’s disease therapies will be developed to prevent the initiation of Alzheimer’s disease.”
“Another important issue is that the necrosis of neurons accumulating intracellular beta-amyloid occurs before formation of beta-amyloid plaques,” continues Professor Okazawa. “Residual beta-amyloid after neuronal necrosis seems to be the seed for beta-amyloid plaques outside of neurons. This discovery might change the amyloid hypothesis considering that extracellular beta-amyloid plaque is the top of pathological cascade of Alzheimer’s disease.”
###
The article, “YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology,” was published in Nature Communications at DOI: 10.1038/s41467-020-14353-6.
Media Contact
Hitoshi Okazawa
[email protected]
Original Source
http://www.
Related Journal Article
http://dx.


{kind=link}