
Prostate cancer remains one of the most prevalent malignancies affecting men worldwide, accounting for a significant burden of cancer incidence, particularly in Europe and North America where it is the leading male cancer diagnosis. Despite substantial advances in therapeutic strategies targeting androgen signaling and the androgen receptor axis, a notable proportion of patients—approximately 30%—progress to advanced stages characterized by metastatic castration-resistant prostate cancer (mCRPC) and neuroendocrine prostate cancer subtypes. These forms are notorious for their aggressiveness and therapeutic resistance, presenting formidable obstacles in clinical oncology. Consequently, the field is urgently seeking novel molecular targets to circumvent resistance mechanisms and improve patient outcomes.
A groundbreaking avenue in prostate cancer research centers around the pioneer transcription factor FOXA1. This factor, frequently mutated in prostate tumors—ranking as the third most mutated gene—plays an indispensable role in the initiation and progression of prostate malignancies. FOXA1’s regulatory function is largely contingent on post-translational modifications; however, the intricate details governing these modifications have remained elusive until now. Parallel to this, cyclin-dependent kinase 12 (CDK12), a kinase with recognized involvement in transcription elongation and DNA damage response, has emerged as a critical player in prostate cancer pathobiology. Genetic aberrations in CDK12 correlate strongly with disease progression and unfavorable prognosis.
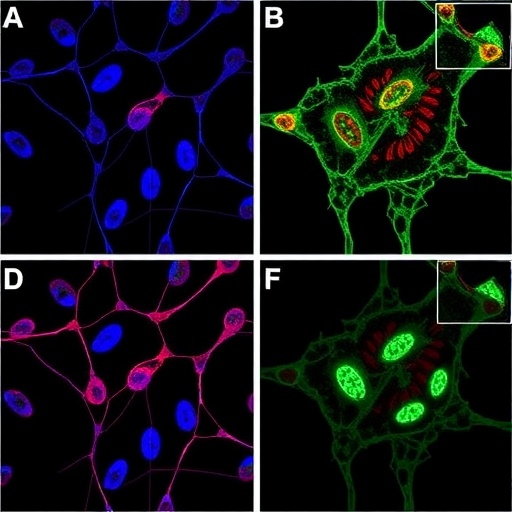
Recent research has for the first time delineated a direct mechanistic link between CDK12 and FOXA1, unveiling a novel signaling axis integral to prostate tumor development. The study identifies CDK12 as a direct kinase for FOXA1, revealing a phosphorylation-dependent activation pathway that propels oncogenic processes. Central to this axis is the phosphorylation of FOXA1 at serine residue 234 (S234), a highly conserved amino acid within the DNA-binding domain of FOXA1, which modulates its transcriptional activity and downstream gene regulatory functions.
The identification of this site was accomplished through sophisticated bioinformatics analyses complemented by rigorous in vitro and in vivo validation experiments. The researchers engineered precise site-directed mutants of FOXA1—S234A to represent a non-phosphorylatable form, and S234E as a phosphomimetic version—thereby enabling detailed functional dissection of this modification. Crucially, the development of a novel, site-specific antibody against phosphorylated S234-FOXA1 furnished a powerful tool for probing the dynamics of this modification in cellular contexts.
Mechanistically, this phosphorylation event amplifies FOXA1’s chromatin binding affinity and transcriptional potency without altering its cellular localization. Functional genomics and reporter assays illuminated that phosphorylated FOXA1 directly upregulates MDM2, an E3 ubiquitin ligase that orchestrates the ubiquitination and subsequent proteasomal degradation of the tumor suppressor p53. By intensifying MDM2 transcription, phosphorylated FOXA1 effectively diminishes p53 protein stability, thereby suppressing apoptosis and fostering a cellular milieu conducive to cancer cell survival and unchecked proliferation.
The CDK12-FOXA1-MDM2-p53 signaling cascade represents a comprehensive oncogenic axis in prostate cancer. Disruption of this pathway, particularly at the level of CDK12 catalytic activity, emerges as a promising therapeutic intervention point. The study showcases that THZ531, a selective small molecule inhibitor of CDK12/13, robustly suppresses FOXA1 transcriptional activity and compromises tumor cell viability. Notably, in vivo experiments utilizing prostate cancer xenograft models in immunocompromised mice demonstrated that THZ531 administration significantly retards tumor growth, restores p53 protein levels by reducing MDM2 expression, and curtails malignant progression.
The implications of these findings extend beyond fundamental mechanistic insights. They offer a tangible strategy for tackling subsets of prostate cancer patients characterized by aberrant CDK12 activity or elevated FOXA1 expression. Targeting CDK12 with inhibitors such as THZ531 promises a dual-pronged therapeutic effect: attenuating FOXA1’s oncogenic transcriptional output alongside stabilizing p53, the guardian of the genome, effectively disrupting cancer-promoting signals from multiple angles.
Importantly, the phosphorylation-mediated regulation of FOXA1 outlined in this study enriches the understanding of post-translational modification networks that fine-tune transcription factor function in cancer. It also bridges the gap between FOXA1 and the classical MDM2-p53 tumor suppressor pathway, a relationship previously unrecognized in prostate oncogenesis. This discovery thus anchors FOXA1 not only as a pioneer factor for chromatin remodeling but also as a pivotal modulator of tumor suppressor homeostasis.
While this research solidifies the role of CDK12-driven FOXA1 phosphorylation in apoptosis inhibition and proliferation, it opens new avenues for investigating broader epigenomic ramifications. Future work is warranted to explore how S234 phosphorylation influences genome-wide chromatin plasticity, affects global gene expression patterns, and intersects with androgen receptor signaling pathways, which remain central to prostate cancer biology.
Moreover, clinical translation of these insightful findings is a high priority. Rigorous clinical trials assessing the safety, efficacy, and combinatorial potential of CDK12 inhibitors like THZ531 alongside established therapies—such as androgen deprivation and chemotherapy—will be essential. Such studies may pave the way for personalized medicine approaches that exploit the vulnerabilities of the CDK12-FOXA1-MDM2-p53 axis in treatment-resistant prostate cancers.
Overall, this research marks a significant leap forward in prostate cancer biology and therapeutic development. By illuminating a precise molecular mechanism that drives tumor progression, it provides a robust scientific foundation for new treatment paradigms aimed at improving the prognosis for patients facing advanced, refractory disease.
Subject of Research:
Not applicable
Article Title:
CDK12-Mediated Phosphorylation of FOXA1 Promotes Prostate Cancer Progression via the MDM2–p53 Axis
News Publication Date:
10-Nov-2025
Web References:
http://dx.doi.org/10.34133/research.0990
Keywords:
Prostate cancer, CDK12, FOXA1, phosphorylation, MDM2, p53, transcription factor, tumor progression, post-translational modification, kinase inhibitor, THZ531, apoptosis, chromatin binding
Tags: advanced prostate cancer subtypesandrogen receptor therapy resistanceCDK12-FOXA1 molecular pathwayclinical implications of CDK12cyclin-dependent kinase 12 role in cancerFOXA1 transcription factor mutationsmale cancer incidence trendsneuroendocrine prostate cancernovel molecular targets in oncologypost-translational modifications in tumorsprostate cancer progressiontherapeutic strategies for mCRPC


{kind=link}