Understanding the pathogenesis of hereditary polycystic kidney disease and developing new treatments
Credit: Professor Ryuichi Nishinakamura
Joint research, led by Kumamoto University in Japan, has successfully reproduced the pathogenesis of autosomal dominant polycystic kidney disease (ADPKD) from human iPS cells in vitro. ADPKD is a disease that causes multiple cysts in both kidneys and is the most common hereditary kidney disease. Although cysts derived from renal tubules have been previously documented, this is the first induction of cysts from collecting ducts, which is more closely related to the pathogenesis of the disease. The researchers expect that this will lead to a better understanding of disease states and the development of new treatment methods.
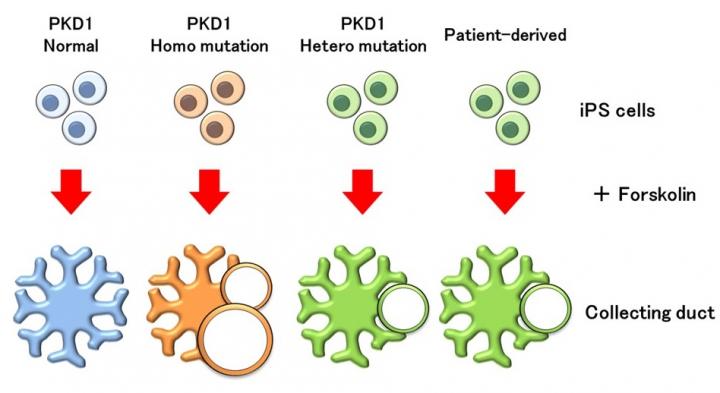
ADPKD is estimated to affect 1 in every 400-1,000 people. Although half of ADPKD patients progress to renal failure by the age of 60, the underlying mechanisms of the disease are not clear and a treatment has yet to be found. ADPKD is caused by mutations in the PKD genes, and will develop if one or both copies of the PKD genes, inherited from the father and mother each, have mutations (heterozygous or homozygous mutations). The PKD genes consist of PKD1 and PKD2 and about 85% of all ADPKD patients have a heterozygous mutation of the PKD1 gene. The remaining 15% have a heterozygous mutation of the PKD2 gene. Those with heterozygous mutations of the PKD1 gene have a more severe prognosis, with approximately half developing renal failure by the age 60 due to the progression of renal cysts. The mechanism of ADPKD has been mainly studied in mice. However, mice with a heterozygous mutation in the PKD1 gene develop few renal cysts, even in adults, and human symptoms are not reproduced.
Kidneys develop through the interactions between the two different precursors; nephron progenitors and ureteric buds. Nephron progenitors differentiate into the renal tubules that reabsorb salt and water in the urine, and ureteric buds differentiate into the collecting ducts that collect urine and reabsorb water. ADPKD cysts can originate from both the renal tubules and the collecting ducts, but the collecting ducts appear to be their predominate origin. In 2014, Nishinakamura et al. reported on a method of inducing renal tubules from human iPS cells via nephron progenitor cells. Since then, several research groups have successfully produced renal tubule-derived cysts from iPS cells that had a homozygous mutation of the PKD1 gene, but cysts derived from collecting ducts have not yet been produced. Because renal tubule-cysts have also been formed from normal iPS cells without gene mutation, it was not possible to reproduce the disease state from iPS cells derived from ADPKD patients who had the heterozygous mutation of the PKD1 gene. However, Professor Nishinakamura’s research group induced collecting ducts from human iPS cells via ureteric buds in 2017, and have since been attempting to reproduce cysts derived from collecting duct by building on their own methods.
To reproduce the ADPKD cysts, the researchers used the gene editing technology CRISPR-Cas9 to create both homozygous and heterozygous mutant iPS cells with the PKD1 gene and induced them into renal tubules. After administration of a drug called forskolin, which activates a factor that exacerbates ADPKD cysts, tubular cysts were reproduced as in previous reports. However, mild cysts also formed from the renal tubules that had not been genetically mutated.
When these iPS cells were induced into collecting ducts and treated with forskolin, cysts formed from the collecting duct that had the PKD1 homozygous mutation. On the other hand, cysts did not form from collecting ducts that had no gene mutations, indicating that their response to forskolin was different from that of renal tubules. When researchers checked the expression of the receptor for the antidiuretic hormone vasopressin, which acts on the collecting duct, they found that it was only expressed in the collecting duct. Vasopressin is known to exacerbate ADPKD cysts from collecting ducts, and when it was administered to the induced collecting ducts and renal tubules, cysts formed, albeit at a low frequency, only in collecting ducts with PKD1 homozygous mutation. Furthermore, cysts partially formed in collecting ducts with the PKD1 heterozygous mutation after administration of forskolin. When iPS cells from an ADPKD patient with the PKD1 heterozygous mutation were used to induce collecting ducts, cysts formed in the same way. According to the researchers, this is the first successful reproduction of ADPKD disease pathology from patient-derived iPS cells.
“This study shows that the method of inducing collecting ducts from iPS cells can be a new disease model for ADPKD,” said study leader, Professor Nishinakamura. “By analyzing these collecting duct cysts–which are similar to actual clinical conditions–we may find mechanisms and develop new therapeutic methods that have been difficult to identify until now. We also expect that the replication of cysts from patient-derived iPS cells will lead to research and treatments for individual cases.”
###
This research was posted online in the Journal of the American Society of Nephrology on 3rd August 2020.
[Source]
Kuraoka, S., Tanigawa, S., Taguchi, A., Hotta, A., Nakazato, H., Osafune, K., … Nishinakamura, R. (2020). PKD1-Dependent Renal Cystogenesis in Human Induced Pluripotent Stem Cell-Derived Ureteric Bud/Collecting Duct Organoids. Journal of the American Society of Nephrology, ASN.2020030378. doi:10.1681/asn.2020030378
Media Contact
J. Sanderson, N. Fukuda
[email protected]
Original Source
http://www.
Related Journal Article
http://dx.



{kind=link}