Accelerating search for various materials to achieve a carbon-free society
Credit: Atsushi Ishikawa
Japanese researchers have developed a simulation method to theoretically estimate the performance of heterogeneous catalyst by combining first-principles calculation (1) and kinetic calculation techniques. Up to now, simulation studies mainly focused on a single or limited number of reaction pathways, and it was difficult to estimate the efficiency of a catalytic reaction without experimental information.
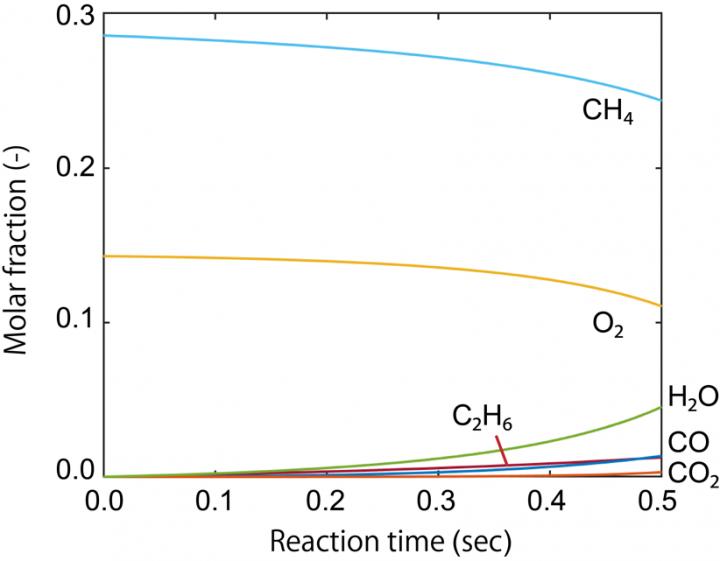
Atsushi Ishikawa, Senior Researcher, Center for Green Research on Energy and Environmental Materials, National Institute for Materials Science (NIMS), performed computation of reaction kinetic information from first-principles calculations based on quantum mechanics, and developed methods and programs to carry out kinetic simulations without using experimental kinetic results. Then he applied the findings to the oxidative coupling of methane (OCM) reaction, which is an important process in the use of natural gas. He could successfully predict the yield of the products, such as ethane, without experimental information on the reaction kinetics. He also predicted changes in yield depending on the temperature and partial pressure, and the results reproduced faithfully the existing experimental results.
This research shows that the computer simulation enables the forecasting the conversion of reactant and the selectivity of products, even if experimental data are unavailable. The search for catalytic materials led by theory and calculation is expected to speed up. Furthermore, this method is highly versatile and can be applied not only to methane conversion catalysts but also to other catalyst systems such as for automobile exhaust gas purification, carbon dioxide reduction and hydrogen generation, and is expected to contribute to the realization of a carbon-free society.
###
The research was supported by JST’s Strategic Basic Research Program, Precursory Research for Embryonic Science and Technology (PRESTO) Program.
(1) First-principles calculation
A theoretical calculation method for solving quantum mechanical equations without using empirical parameters, which is often applied to atomic, molecular, solid, and interface systems.
Media Contact
Atsushi Ishikawa
[email protected]
Original Source
https:/
Related Journal Article
http://dx.



{kind=link}