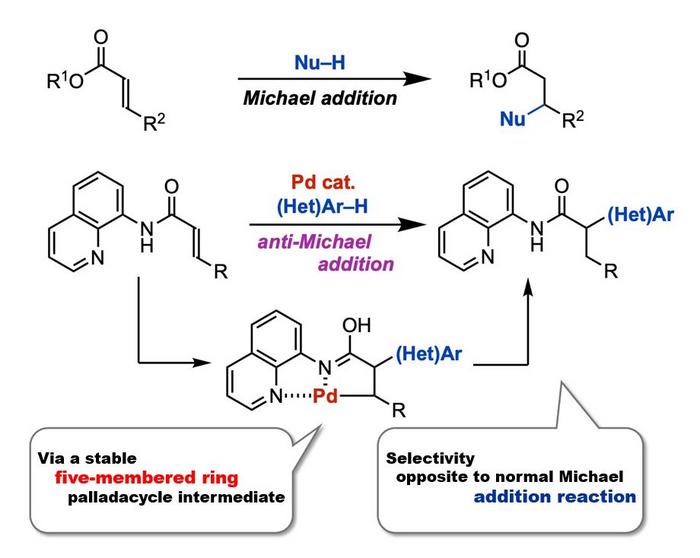
In 1887, chemist Sir Arthur Michael reported a nucleophilic addition reaction to the β-position of α,β– unsaturated carbonyl compounds. These reactions, named Michael addition reactions, have been extensively studied to date. In contrast, the anti-Michael addition reaction, referring to the nucleophilic addition reaction to the α-position, has been difficult to achieve. This is due to the higher electrophilicity of the β-position compared to the α-position. Previous attempts to overcome these difficulties have involved two main methods. The first is restricting the addition position via intramolecular reactions, while the second method involves introducing a strong-electron withdrawing group at the β-position. However, these methods are not ideal for synthesizing complex molecules via the anti-Michael reaction.
Credit: Cannot be reused without permission
In 1887, chemist Sir Arthur Michael reported a nucleophilic addition reaction to the β-position of α,β– unsaturated carbonyl compounds. These reactions, named Michael addition reactions, have been extensively studied to date. In contrast, the anti-Michael addition reaction, referring to the nucleophilic addition reaction to the α-position, has been difficult to achieve. This is due to the higher electrophilicity of the β-position compared to the α-position. Previous attempts to overcome these difficulties have involved two main methods. The first is restricting the addition position via intramolecular reactions, while the second method involves introducing a strong-electron withdrawing group at the β-position. However, these methods are not ideal for synthesizing complex molecules via the anti-Michael reaction.
In a new study, a global team of researchers, led by Professor Takanori Matsuda and including Mr. Ryota Moro, both from the Department of Applied Chemistry at Tokyo University of Science, Japan, as well as including Assistant Professor Hirotsugu Suzuki from the Tenure-Track Program for Innovative Research at the University of Fukui, Japan, successfully achieved palladium-catalyzed anti-Michael addition reaction of acrylamides. This represents the first example of an anti-Michael-type addition reaction. “We found that the presence of a catalytic amount of palladium(II) trifluoroacetate Pd(TFA)2 is capable of facilitating the anti-Michael addition of indole to acrylamide with an aminoquinoline group as a directing group, producing the addition product in high-yield,” explains Prof. Matsuda.
Their study was made available online on May 14, 2024, and published in Volume 146, Issue 20 of the Journal of the American Chemical Society on May 22, 2024.
The team reasoned introducing a directing group into an α,β-unsaturated carbonyl compound could facilitate an anti-Michael type addition reaction by stabilizing the reaction intermediate. To test this, the researchers first used an acrylamide having an aminoquinoline-directing group, and a nucleophile, 1-methylindole, as model substrates to investigate the anti-Michael type addition reaction in the presence of the palladium catalyst. This reaction produced the desired product with a 90% yield. At a reaction scale of two millimoles, there was no yield loss, signifying the practicality of the reaction.
This reaction was also carried out with β-substituted cinnamamide derivatives and crotonamide derivatives with an alkyl group. Moreover, the reaction proceeded smoothly with a wide range of nucleophiles, including many indoles, heterocyclic compounds such as pyrroles and thiophenes, and electron-rich aromatic compounds. Additionally, the aminoquinoline-directing group used in this reaction can be converted to carboxylic acids and other amides, signifying the usefulness of the reaction.
The researchers also investigated the mechanism for this reaction through labeling experiments. They found that initially, the acrylamide coordinates to Pd(TFA)2 to form a five-membered ring palladacyle intermediate. The reaction then proceeds with the nucleophilic attack by indole on the intermediate, producing alkylpalladium species. Finally, an acid removes palladium and regenerates Pd(TFA)2, producing the desired α-substituted carbonyl compound.
Highlighting the potential applications of this study, Dr. Suzuki says, “The anti-Michael type addition is expected to become an ideal one-step reaction with 100% atomic efficiency for the synthesis of α-substituted carbonyl compounds, which are often used in pharmaceuticals. Our method will enable the widespread application of this reaction.”
Overall, this novel method can lead to efficient and sustainable synthesis of α-substituted carbonyl compounds and consequently pharmaceuticals, among other organic compounds.
***
Reference
DOI: https://doi.org/10.1021/jacs.4c00841
Authors: Hirotsugu Suzuki1, Ryota Moro2, and Takanori Matsuda2
Affiliations:
1Tenure-Track Program for Innovative Research, University of Fukui, Japan
2Department of Applied Chemistry, Tokyo University of Science, Japan
Funding information
This work was supported by JSPS KAKENHI Grant Numbers JP23K13743 and JP21K05061.
Journal
Journal of the American Chemical Society
DOI
10.1021/jacs.4c00841
Method of Research
Experimental study
Subject of Research
Not applicable
Article Title
Palladium-Catalyzed anti-Michael-Type (Hetero)arylation of Acrylamides
Article Publication Date
22-May-2024
COI Statement
The authors declare no competing financial interest.

{kind=link}