Preclinical study identifies vulnerability in orphan cancers with SMARCB1 mutations, spurs launch of Phase II clinical trial

Credit: MD Anderson Cancer Center
Researchers at The University of Texas MD Anderson Cancer Center have discovered that malignant rhabdoid tumors (MRT), a rare pediatric cancer without effective treatments, may be sensitive to drugs that block the cancer cell’s ability to dispose of misfolded proteins. The findings provide a much-needed therapeutic target for these and other cancers caused by mutations in the SMARCB1 gene.
Based on these findings, published today in Cancer Cell, the researchers now are leading a Phase II clinical trial to investigate this approach in renal medullary carcinoma (RMC), a related adolescent cancer also characterized by SMARCB1 mutations.
Malignant rhabdoid tumors are aggressive pediatric cancers that generally manifest in children in the first year of life, explains Giannicola Genovese, M.D., assistant professor of Genitourinary Medical Oncology and corresponding author on the study.
“It’s somewhat fortunate that these are very rare cancers. With current treatments, the prognosis is ominous – just about one year,” said Genovese. “It’s an orphan disease, meaning it has no targeted treatments available. The current options include high-dose chemotherapy, radiation and surgery, which can cause significant developmental and neurological effects and increase the risk for secondary cancers later in life.”
Driven to answer the unmet needs for patients with MRT and other cancers caused by SMARCB1 mutations, the researchers developed a mouse model of MRT to study the disease biology and uncover vulnerabilities. The researchers created a mosaic mouse model, meaning only a subset of cells carried the mutation, to study the loss of SMARCB1 in specific cells and tissues in the mice.
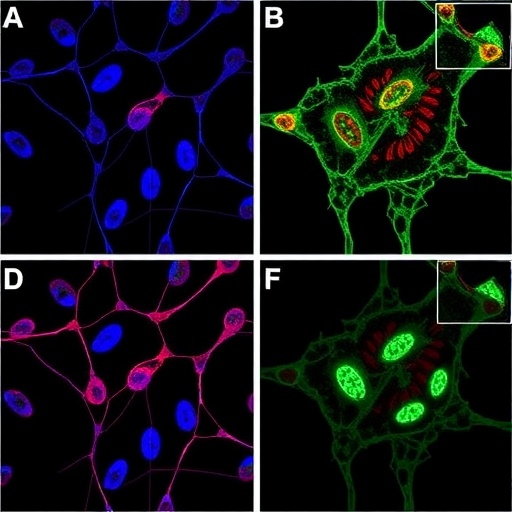
“Using an embryonic mosaic approach, we were able to overcome current limitations in technology and create a model that faithfully mimics features of human MRT,” said Alessandro Carugo, Ph.D., institute research scientist at the Institute for Applied Cancer Science (IACS). “We demonstrated, in our model, that SMARCB1-deficient cells are characterized by dysregulation of protein synthesis and disposal, causing them to be loaded with misfolded protein aggregates.”
Excess or misfolded proteins can be toxic to the cell, so the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress response pathways are normally activated to clear protein waste. However, with loss of SMARCB1, these pathways were hijacked and incorrectly activated to account for protein accumulation in malignant cells.
The researchers also discovered that the cancer cells were able to survive this buildup through the activity of p53, a tumor suppressor that normally acts to block tumor growth. In contrast, SMARCB1-deficient cells appear to activate p53 for a survival advantage and, in fact, may benefit from chemotherapy treatments, which further stimulate p53 activity.
“TP53 is the most frequently mutated gene in cancer, but it does not appear to be mutated in MRT,” said Carugo. “We were excited to learn that, here, it’s functional because the cells need p53 to counterbalance the protein accumulation associated with SMARCB1 loss.”
Recognizing the mis-regulated protein pathways as a potential vulnerability, the researchers leveraged the capabilities of MD Anderson’s Therapeutics Discovery platforms, including IACS, to design a drug combination to test this hypothesis. The Therapeutics Discovery division is a unique group of clinicians, researchers and drug discovery scientists working to develop innovative treatment options, inspired by the needs of patients and guided by the expertise of MD Anderson physicians.
The researchers targeted two mechanisms important for clearing protein waste, known as autophagy and proteasome degradation. Blocking these in the mouse model led to complete tumor regression, and the results were further confirmed in mouse transplants and cell lines derived from human MRT.
The findings of this study led the researchers to initiate a Phase II clinical trial testing a proteasome inhibitor in combination with chemotherapy for patients with RMC. This rare form of kidney cancer, also driven by SMARCB1 mutations, affects adolescents and young adults, primarily African Americans with the sickle-cell trait. The trial also is available for adolescents and adults who develop MRT in their kidney.
“This is the first trial designed specifically for SMARCB1-mutant RMC. These are rare cancers that don’t respond to the standard of care for other kidney cancers,” said Genovese. “There is a need for new therapies for treating these patients. I think we’ve developed a good platform for discovery, to provide options for these patients and hope for the families as well.”
The researchers are working to expand the trial in the future to include pediatric MRT and other SMARCB1-related cancers. In addition, the Therapeutics Discovery team will continue to investigate new medicines that may benefit these patients in the future.
###
Genovese is corresponding author of the study together with Nizar Tannir, M.D., professor of Genitourinary Medical Oncology, and Giulio F. Draetta, M.D., Ph.D., chief science officer ad interim and senior vice president of Therapeutics Discovery. A complete list of authors involved with the study can be found with the full article. The authors declare no conflicts of interest.
The study was supported by the Conquer Cancer Foundation Young Investigator Award, the Kidney Cancer Association Young Investigator Award, the Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Research, the Pancreatic Cancer Action Network, the Italian Foundation for Cancer Research and Italian Association for Cancer Research (FIRC-AIRC), the Ransom Horne, Jr. Professorship for Cancer Research, the Barbara Massie Memorial Fund and the Cancer Prevention and Research Institute of Texas (RP170722).
Media Contact
Clayton R. Boldt, Ph.D.
[email protected]
713-792-9518
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}