A team of scientists from Sechenov First Moscow State Medical University (MSMU) together with their colleagues described a number of genetic mutations causing Usher syndrome (inherited deaf-blindness)
Credit: Courtesy of Dmitry Zaletaev
A team of scientists from Sechenov First Moscow State Medical University (MSMU) together with their colleagues from leading scientific centers of Moscow and India described a number of genetic mutations causing Usher syndrome (inherited deaf-blindness). They found previously unstudied unique mutations in investigated DNA regions. The results of the study were published in the Ophthalmic Genetics journal.
Usher syndrome is an inherited disorder that accounts for almost a half of all cases of deaf-blindness. There are three types of the disease that are differentiated based on the level of deafness (type I – innate deafness, type II – diminished hearing that does not worsen with time, and type III -progressive decrease in hearing). According to one of the works dating back to 2010, the disease affects 3 to 6 in 100,000 people depending on the region of study. However, among American children the share of patients with this condition may reach 1 in 6,000. Mutations that cause Usher syndrome occur with different frequencies in different populations.
The authors of the work studied and described mutations in Russian patients with Usher syndrome. Due to the rarity of this condition the team worked with 28 patients (11 men and 17 women) 35-60 years of age in which the disease manifested when they were 1 to 18 years old. The group consisted of 21 (75%) Russians (of Slavic origin), 3 (10%) Ukrainians, 2 (7%) Jews, 1 (4%) Belarusian, and 1 (4%) Chuvash. The patients were selected according to their medical records and underwent tests to confirm their clinical diagnosis. All participants suffered from vestibular sickness and vision disorders, and 22 out of 28 also had hearing problems.
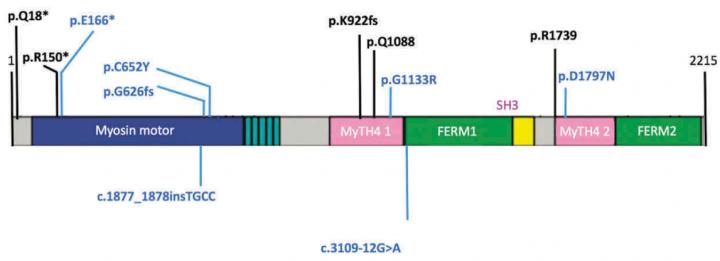
Clinical examinations of vision, hearing, and vestibular system showed that 15 and 11 patients had USH of type I and II respectively. Genetic testing demonstrated different mutations associated with the development of Usher syndrome in 23 patients. 11 patients had mutations in the genes associated with type I USH: MYO7A (8 people; a gene that codes a protein in charge of cell movement and intracellular transportation), CDH23 (1), PCDH15 (1) (genes that code signal proteins playing a role in the work of retina and inner ear), and USH1C (1) (a gene that codes a protein playing a role in the development of retina and inner ear). MYO7A displayed 11 mutations, 6 of them have not been described before. 2 new mutations were found in the PCDH15 gene. Genetic testing confirmed around 91% of type II USH diagnoses. The USH2A gene associated with type II USH showed 11 mutations, 3 of which turned out to be new. The prevalence of type II USH in Russian patients was lower than in other countries, and the frequency of mutations in MYO7A in charge of type I USH was very high.
“While analyzing the databases of mutations associated with Usher syndrome we understood that some of the mutations we had found were completely new. Unfortunately, they would not help recognize the disease on earlier stages or with higher probability, but would be able to confirm the diagnosis. The search for new genetic variations associated with the syndrome may help identify new targets for specific therapy. However, this is not happening in the near future,” said Prof. Dmitry Zaletaev, a chief research associate at the laboratory of Medical Genetics, Sechenov University.
###
Among the participants of the study were scientists from India, employees of Ophthalmic Ltd, Center for Ophthalmology of the Federal Medical and Biological Agency of Russia, Research Centre for Medical Genetics, Institute of General Pathology and Pathophysiology, and many others. The work was carried out with the support of So-Edinenie Deaf-Blind Support Foundation and Sensor-Tech Laboratory.
Media Contact
Nataliya Rusanova
[email protected]
Original Source
https:/
Related Journal Article
http://dx.


{kind=link}