New molecular insights could mean more accurate prognoses and better selection of therapies
Credit: Acta Neuropathologica Communications
Researchers at the Johns Hopkins Kimmel Cancer Center report that their study of tumor samples from people with the rare genetic syndrome neurofibromatosis type 1 (NF1) has uncovered novel molecular clues about which tumors are most likely to be aggressive in those with NF1. According to the researchers, the clues could advance the search for more customized and relevant treatments that spare patients exposure to treatments unlikely to work.
“Some NF1 tumors are hard to classify as benign or malignant,” says study leader Fausto Rodriguez, M.D., associate professor of pathology, oncology, and ophthalmology at the Johns Hopkins University School of Medicine and a member of the Johns Hopkins Kimmel Cancer Center. “Having another molecular marker to evaluate will help us make better predictions about the outcomes of treatment.”
A summary of the study was published online in the journal Acta Neuropathologica Communications on Aug. 28.
NF1 is caused by mutations in the NF1 gene. Although rare overall, occurring in about one in 3,000 people, it is among the most common so-called genetic syndromes. About half of all cases are inherited.
People with NF1 are predisposed to develop multiple tumors in the brain and along peripheral nerves throughout the body, just under the skin, and also are predisposed to form rarer tumors of the gastrointestinal tract, adrenal gland and other soft tissues. The overall number of tumors per patient varies greatly, from several to hundreds over their lifetimes. While most are slow-growing (low-grade) and benign, some benign peripheral nerve tumors (neurofibromas) develop into so-called malignant peripheral nerve sheath tumors (MPNST), which metastasize frequently, are challenging to treat and are a major cause of death in patients with NF1.
Over time, Rodriguez said he noticed that many of the high-grade, aggressive NF1 tumors he examined were lacking the protein ATRX, a loss associated with a cancer-specific biological process called ALT, short for alternative lengthening of telomeres. Co-investigator Christopher Heaphy, Ph.D., ALT expert and assistant professor of pathology and oncology at Johns Hopkins, explains that at the end of each chromosome in a cell’s nucleus, a sequence of six DNA “letters” is repeated up to 2,000 times to form a so-called telomere. “Telomeres are like the plastic caps on the ends of shoelaces,” he says. “They protect chromosomes from fraying or getting jumbled.”
Before a cell divides, it must duplicate its chromosomes, along with their telomeres, but the very tips of the telomeres can’t be replicated. So a few dozen DNA letters are lost each time a cell divides, making telomere length a stand-in for the age of a cell, says Heaphy.
Since cancer cells divide more frequently than most healthy cells, their telomeres shorten more quickly, putting the cell in danger of chromosomal instability, cell death and “genomic catastrophe,” says Heaphy. To combat this, most cancer cells turn on a special enzyme called telomerase to maintain the length of their telomeres. (Carol Greider, Ph.D., the Daniel Nathans Professor and Director of Molecular Biology and Genetics at Johns Hopkins, won the 2009 Nobel Prize in Physiology or Medicine for her discovery of telomerase.)
“A subset of cancers, however, do not activate telomerase but use the ALT mechanism instead,” says Heaphy. ALT uses homologous recombination, a type of exchange of genetic material, to lengthen telomeres, but how exactly this happens remains under investigation.
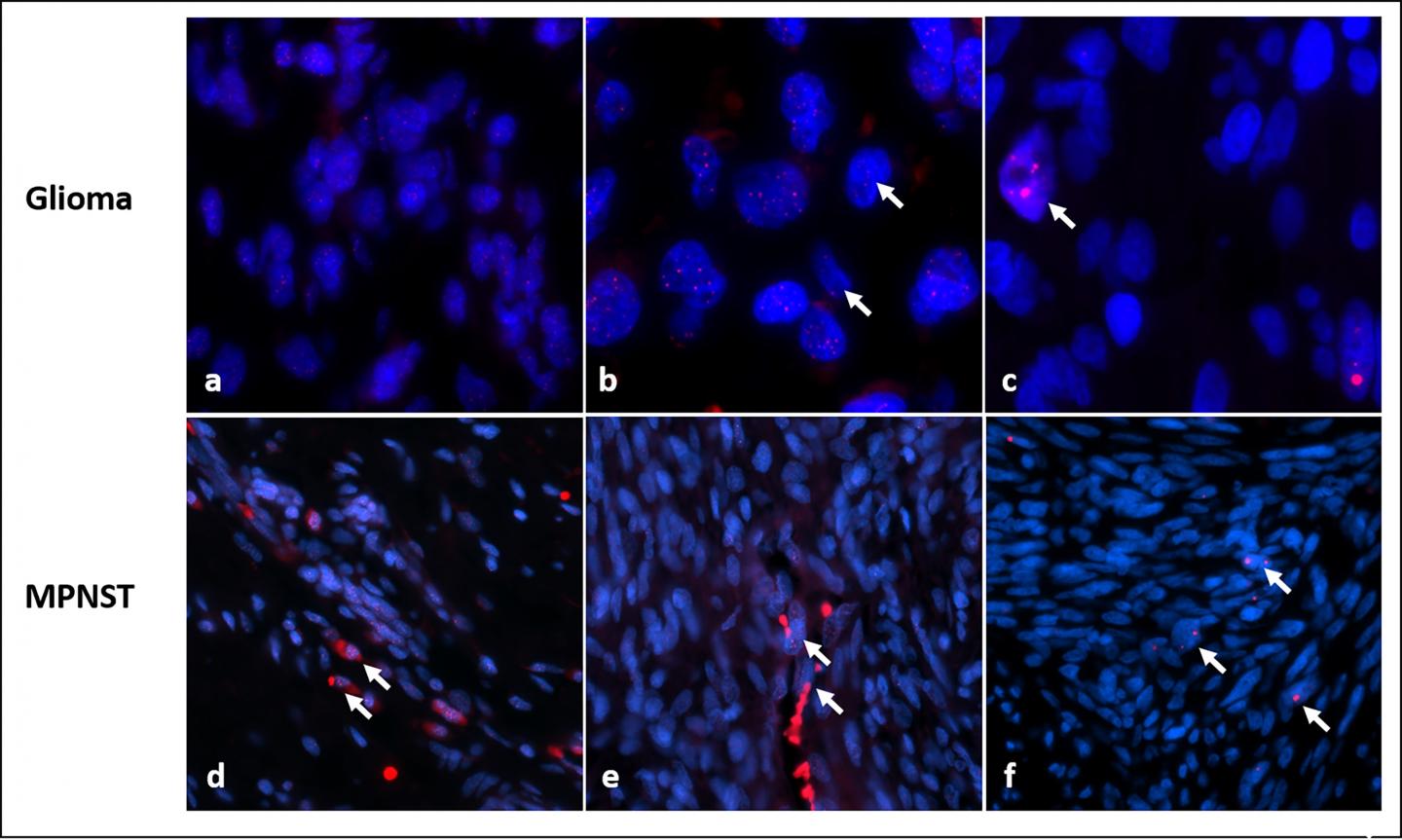
To assess the importance of ALT in NF1 tumors, Rodriguez, Heaphy and colleagues used fluorescent probes that illuminated the telomeres to examine 426 tumor samples from 256 patients with NF1.
Overall, ALT was detected in 23 NF1-associated gliomas (32.9%). Of the ALT-positive tumors, 14 (60%) were high-grade, or aggressive, tumors, while only 9 of 47 low grade gliomas (19%) were ALT positive.
Among the 99 MPNST samples, 93 developed in patients known to have NF1. ALT was present in 15 of 148 (17%) patients, but ALT was almost entirely absent from the 77 neurofibromas studied and all other rarer tumors (46), except for two cases, which were both malignant.
“The data show that ALT is usually — although not always — associated with more aggressive tumors in patients with NF1,” says Rodriguez. “It’s also more likely to occur in gliomas than MPNSTs.”
Using the same assay, the team also assessed cells from tumors that were ALT-negative. They assessed the overall telomere lengths and compared them with those in non-cancerous cells within the same sample, and then scored as short, normal or long.
In the 183 ALT-negative neurofibromas assessed, all the telomere lengths were considered normal. By contrast, normal telomeres were found in only 57% and 50%, respectively, of the 30 ALT-negative gliomas and 30 ALT-negative MPNSTs tested. While altered telomere lengths were prevalent, abnormal telomeres tended to be too long in gliomas (40%), and too short in MPNSTs (47%).
Rodriguez says those differences could be used clinically to predict tumor aggressiveness, but he cautioned that the differences also could stem from variations in patient age, the fact that standard telomere lengths differ in various cell types, the timing of telomerase activation, or something else. Larger studies are needed, he adds, to rule out those possibly confounding factors.
Finally, the researchers analyzed correlations between telomere alterations and patient survival. Patients with gliomas survived longer if they were ALT-negative even when adjusting for age and grade, two important factors affecting outcome in brain tumors. For patients with MPNSTs there was no statistically significant difference between those that were ALT-positive or negative. However, among the whole group of MPNSTs, the researchers found that short telomeres appeared to provide a significant survival advantage, a finding they called “counterintuitive” and that remains unexplained.
“While ALT analysis is not yet a clinical test, surrogate markers such as ATRX loss, may be useful in NF1 patients as a prognostic indicator of more aggressive tumors and worse survival,” says Rodriguez.
###
In addition to Rodriquez and Heaphy, other researchers included Mindy Graham, Jacqueline Brosnan-Cashman, John Barber, Christine Davis, M. Adelita Vizcaino, Doreen Palsgrove, Caterina Giannini, Melike Pekmezci, Sonika Dahiya, Murat Gokden, Michael Noë, Laura Wood, Christine Pratilas, Carol Morris, Allan Belzberg and Jaishri Blakeley.
The research was funded by Pilocytic/Pilomyxoid Research Fund, including Lauren’s First and Goal, and the Stick it to Brain Tumors Annual Women’s Ice Hockey Tournament; the Basic/Translational Science Investigator Award from the North American Neuroendocrine Tumor Society supported by the Neuroendocrine Tumor Research Foundation; a collaborative grant between the Wilmer Eye Institute and the King Khaled Eye Specialist Hospital in Riyadh, Saudi Arabia; Department of Defense grant W81XWH-18-1-0496; and National Institutes of Health grants 2T32CA009110-39A1 and P30 CA006973.
Media Contact
Amy Mone
[email protected]
410-614-2915
Original Source
https:/
Related Journal Article
http://dx.


{kind=link}