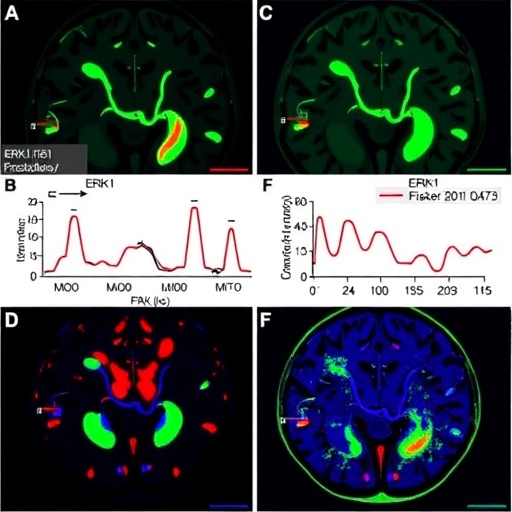

In the relentless pursuit to understand the metabolic adaptations enabling glioblastoma cells to survive in harsh tumor microenvironments, recent groundbreaking research has unveiled a pivotal mechanism that sustains tumor viability under extreme glucose deprivation. The study, conducted by Li, Y., Zhang, F., Hu, F., and colleagues, presents compelling evidence that ERK1-mediated phosphorylation of GLYCTK2 significantly promotes fructolysis—a metabolic detour that glioblastoma cells exploit to maintain their energy demands when glucose supply plummets.
Glioblastoma, notorious for its aggressive clinical course and resistance to treatment, often faces fluctuating nutrient availability within the tumor mass, particularly glucose scarcity. Tumor cells’ metabolic plasticity enables them to switch from glycolysis to alternative pathways to continue generating ATP, the cellular energy currency. This study illuminates how glioblastoma cells harness fructolysis, facilitated by a molecular switch involving ERK1 kinase activity on GLYCTK2, to thrive despite nutrient stress.
ERK1, a mitogen-activated protein kinase, has long been implicated in diverse cell signaling cascades governing proliferation and survival. However, its direct involvement in metabolic regulation within glioblastoma cells presents a novel dimension to its functionality. The phosphorylation of GLYCTK2 — a kinase responsible for key regulatory steps in fructose metabolism — by ERK1 effectively activates fructolysis, enabling glioblastoma cells to bypass glucose dependency.
.adsslot_783tvyIxWB{width:728px !important;height:90px !important;}
@media(max-width:1199px){ .adsslot_783tvyIxWB{width:468px !important;height:60px !important;}
}
@media(max-width:767px){ .adsslot_783tvyIxWB{width:320px !important;height:50px !important;}
}
ADVERTISEMENT
Mechanistically, this phosphorylation event enhances GLYCTK2 enzymatic activity, accelerating the breakdown of fructose into metabolites that feed into the glycolytic and pentose phosphate pathways. Such metabolic reprogramming allows the tumor cells to maintain biosynthetic and bioenergetic homeostasis under conditions where extracellular glucose is insufficient. The research uncovers that this adaptation is not a passive response but an actively regulated survival tactic orchestrated by oncogenic signaling pathways.
To probe this paradigm, the researchers employed a sophisticated combination of phosphoproteomics, in vitro kinase assays, and metabolic flux analysis. These methods collectively validated that GLYCTK2 phosphorylation sites were directly targeted by ERK1, resulting in augmented enzyme functionality. Notably, glioblastoma cell lines subjected to glucose deprivation exhibited elevated levels of phosphorylated GLYCTK2 and concomitant increases in fructose utilization, underscoring the physiological relevance of this regulatory mechanism.
Interestingly, when ERK1 signaling was pharmacologically inhibited or when GLYCTK2 phosphorylation sites were genetically mutated, glioblastoma cells demonstrated impaired fructolysis and reduced viability under low-glucose conditions. This finding not only reinforces the indispensable role of the ERK1-GLYCTK2 axis in metabolic adaptation but also highlights a potential vulnerability for therapeutic exploitation.
The study’s metabolic tracing data further reveal that fructolysis intermediates replenish pools of key metabolites such as glyceraldehyde-3-phosphate and dihydroxyacetone phosphate, feeding into energy-producing and biosynthetic pathways essential for maintaining tumor growth and oxidative balance. This metabolic flexibility grants glioblastoma cells a competitive edge in nutrient-poor microenvironments, facilitating sustained proliferation despite metabolic stress.
Moreover, the investigation delves into the broader implications of fructolysis activation in oncogenic metabolism, suggesting that this pathway may represent a universal adaptation beyond glioblastoma. Since high fructose consumption is increasingly scrutinized for its links to cancer and metabolic disorders, understanding cellular fructose metabolism mechanisms could reveal new intersections between diet, tumor biology, and therapeutic strategies.
The research also posits that ERK1-mediated GLYCTK2 phosphorylation might serve as a biomarker for metabolic phenotyping in glioblastoma, enabling the identification of tumors reliant on fructose metabolism. Such insights pave the way for designing precision medicine approaches that selectively target metabolic dependencies unique to tumor cells, sparing normal tissues that predominantly utilize glucose.
From a translational perspective, the study propounds that small molecule inhibitors disrupting ERK1 activity or GLYCTK2 function could synergize with existing therapies to thwart glioblastoma survival pathways under metabolic duress. Given the notoriously poor prognosis associated with glioblastoma, strategies that cripple the tumor’s metabolic resilience hold promise to improve patient outcomes.
Furthermore, the findings challenge the conventional perception that glucose is the sole major fuel for cancer cells, underscoring the metabolic versatility that underlies tumor progression and therapy resistance. This paradigm shift encourages the scientific community to re-evaluate metabolic targets within the broader context of tumor microenvironmental fluctuations.
In conclusion, this pioneering work elucidates a critical biochemical axis whereby ERK1 orchestrates metabolic adaptation through GLYCTK2 phosphorylation, enabling glioblastoma cells to exploit fructolysis for survival in glucose-deprived niches. The convergence of oncogenic signaling with metabolic reprogramming underscores a sophisticated survival network that cancer cells deploy, offering fresh avenues for therapeutic intervention in one of the most formidable brain tumors.
As the field advances, further exploration into the interplay between kinase signaling and metabolic enzyme regulation will be paramount. Understanding how glioblastoma and other malignancies coordinate such adaptive responses could catalyze the development of next-generation metabolic inhibitors, fine-tuned to disrupt tumor metabolism without harming normal cell function.
This research, published in Cell Death Discovery, not only enriches our fundamental understanding of glioblastoma biology but also propels the development of innovative, metabolism-centered treatment modalities. It stands as a testament to the critical importance of interrogating tumor metabolism within the multifaceted landscape of cancer cell survival strategies.
Subject of Research:
Article Title:
Article References: Li, Y., Zhang, F., Hu, F. et al. ERK1-mediated GLYCTK2 phosphorylation promotes fructolysis to sustain glioblastoma survival under glucose deprivation. Cell Death Discov. 11, 266 (2025). https://doi.org/10.1038/s41420-025-02544-3
Image Credits: AI Generated
DOI: https://doi.org/10.1038/s41420-025-02544-3
Keywords:
Tags: alternative energy pathways in tumorsATP generation under glucose scarcityERK1 phosphorylation in glioblastomafructolysis in cancer metabolismglioblastoma resistance mechanismsglucose deprivation and tumor survivalGLYCTK2 function in glioblastomakinase signaling in cancer metabolismmetabolic adaptations in glioblastomametabolic plasticity of tumor cellsmitogen-activated protein kinase in cancernutrient stress response in glioblastoma

{kind=link}