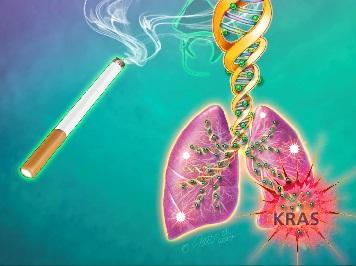
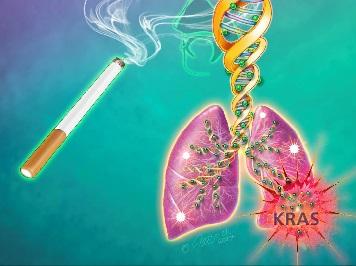
Credit: Jennifer Fairman
Scientists at the Johns Hopkins Kimmel Cancer Center say they have preliminary evidence in laboratory-grown, human airway cells that a condensed form of cigarette smoke triggers so-called "epigenetic" changes in the cells consistent with the earliest steps toward lung cancer development.
Epigenetic processes are essentially switches that control a gene's potentially heritable levels of protein production but without involving changes to underlying structure of a gene's DNA. One example of such an epigenetic change is methylation — when cells add tiny methyl chemical groups to a beginning region of a gene's DNA sequence, often silencing the gene's activation.
"Our study suggests that epigenetic changes to cells treated with cigarette smoke sensitize airway cells to genetic mutations known to cause lung cancers," says Stephen Baylin, M.D., the Virginia and D.K. Ludwig Professor for Cancer Research and professor of oncology at the Johns Hopkins Kimmel Cancer Center. Details of the scientists' experiments are described in the Sept. 11 issue of Cancer Cell.
For two decades, scientists have known some of the genetic culprits that drive lung cancer growth, including mutations in a gene called KRAS, which are present in one-third of patients with smoking-related lung cancers, according to Baylin. Genetic and epigenetic changes also occur when normal cells undergo chronic stress, such as the repeated irritation and inflammation caused by decades of exposure to cigarette smoke and its contents.
Baylin and Johns Hopkins scientist Michelle Vaz, Ph.D., first author on the study, suspected that the interplay of epigenetic and genetic changes may occur when normal lung cells develop into cancer, but, Baylin says, the timing of such changes was unknown.
To create the effect of tobacco smoke on cells, Vaz, Baylin and their colleagues began their studies with human bronchial cells, which line the airways of the lungs, and grew them in a laboratory. Every day for 15 months, the scientists bathed the cells with a liquid form of cigarette smoke, which they say is comparable to smoking one to two packs of cigarettes daily.
The scientists recorded the molecular and genetic changes in the smoke-exposed cells over 10 to 15 months, which the scientists say may be similar to 20 to 30 years of smoking, and compared the changes to bronchial cells that had not been exposed to the liquid smoke.
After 10 days of smoke exposure, the scientists found an overall increase in DNA damage responses to so-called reactive oxygen species within the cells. Reactive oxygen species, also called free radicals, are chemicals that typically contain oxygen, are known to be found in cigarette smoke, and cause DNA damage in cells.
Between 10 days and three months, the cells exposed to smoke had a two- to four-fold increase in the amount of an enzyme called EZH2, which works to dampen the expression of genes. Baylin and other scientists have shown that EZH2 and its effects can precede abnormal DNA methylation in gene start sites.
After EZH2 enzymes rise, their levels taper off, and then, the scientists found two to three-fold increases in a protein called DNMT1, which maintains DNA methylation in the "start" location of a variety of tumor suppressor genes that normally suppress cell growth. When these genes are silenced a barrier is removed that might otherwise stop the cells from growing uncontrollably — a hallmark of cancer.
A host of other genes, which control many other cellular processes do not show such abnormal DNA methylation after smoke exposure.
Baylin says certain genes that control cell growth get turned down periodically during certain stages of life, including embryogenesis, when organisms are growing and developing rapidly. These genes can normally be turned on when cells need to stop growth and allow cells to mature. Chronic cigarette smoke exposure, as noted in many human cancers, tends to block these cell maturation genes from properly turning on, says Baylin.
At the end of six months, the amount of EZH2 and DNMT1 enzymes had tapered off in the cells exposed to the smoke. However, the impact of the two methylation-regulating enzymes was still seen at 10 to 15 months, when scientists found decreased expression of hundreds of genes — many of which are key tumor suppressor genes such as BMP3, SFRP2 and GATA4 — in the smoke-exposed cells and a five- or-more-fold increase in the signaling of the KRAS oncogene that is known to be mutated in smoking-related lung cancers.
However, no mutations were found in the KRAS gene itself or the tumor suppressor genes during the 15-month period of cigarette smoke exposure. These abnormally methylated and silenced genes, says Baylin, would have blocked the increase in KRAS signaling if the genes had been properly activated under smoke-free circumstances.
The scientists also found that the timing of epigenetic and genetic events may be key to lung cancer development. They tested this by inserting mutations into the KRAS gene in the DNA of cells exposed to the cigarette smoke condensate for six months as well as those exposed for 15 months. The scientists found that the inserted mutation transformed cells into cancer in only the 15-month cells, where methylation was fully established, but not in the six-month-exposed cells.
Vaz and Baylin say the results suggest that early epigenetic changes triggered by chronic cigarette smoke exposure can build up over time and make the airway cells increasingly sensitive to responding to mutations that initiate cancer.
They say that smokers can best lower their risk of cancer by quitting altogether, and the sooner a smoker quits, the lower their lung cancer risk may be. Their analysis of data in previous studies done by The Cancer Genome Atlas group have shown that the types of abnormal methylation levels they found are lower in smokers who have quit for more than 10 years than those who have not quit.
It may be possible to use de-methylating drugs, they say, for people with higher than normal risk for lung cancer, such as people who have had surgery for early forms of the disease. Such drugs are currently used in clinical trials for certain types of cancer and are standard therapy for a type of pre-leukemia condition.
The scientists caution that their model, as is the case with any laboratory model, may not be exactly what occurs in people during a lengthy period of smoking, but they say it's a first step in understanding the epigenetic processes that may occur early in the transformation of cells into lung cancer.
The scientists also do not know if their model applies to people who smoke e-cigarettes or other forms of tobacco, as their study used condensates typically found in traditional cigarettes.
###
In addition to Baylin and Vaz, other scientists involved in the research include Stephen Y. Hwang, Ioannis Kagiampakis, Ashwini Patil, Jillian Phallen, Heather M. O'Hagan, Lauren Murphy, Cynthia A. Zahnow, Edward Gabrielson, Victor E. Velculescu and Hariharan P. Easwaran from Johns Hopkins; and Heather M. O'Hagan from the Indiana University School of Medicine.
Michelle Vaz, Stephen Y Hwang, Ioannis Kagiampakis, Ashwini Patil, Jillian Phallen, Heather M O'Hagan, Lauren Murphy, Cynthia A Zahnow, Edward Gabrielson, Victor E Velculescu and Hariharan P Easwaran from Johns Hopkins; and Heather M O'Hagan from the Indiana University School of Medicine.
The research was funded by the National Institutes of Health's National Cancer Institute (CA043318, CA121113, CA006973, CA180950) and the National Institute of Environmental Health Sciences (ES011858, ES023183), the Hodson Trust, the Commonwealth Foundation, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the SU2C-DCS International Translational Cancer Research Dream Team Grant, and the Flight Attendant Medical Research Institute.
Velculescu is a founder of Personal Genome Diagnostics. He is a member of its scientific advisory board and board of directors, and he owns Personal Genome Diagnostics stock, which is subject to certain restrictions under university policy.
Media Contact
Vanessa Wasta
[email protected]
410-614-2916
@HopkinsMedicine
http://www.hopkinsmedicine.org

{kind=link}