Credit: University of Pennsylvania
The immune system is contantly performing surveillance to detect foreign organisms that might do harm. But pathogens, for their part, have evolved a number of strategies to evade this detection, such as secreting proteins that hinder a host's ability to mount an immune response.
In a new study, a team of researchers led by Igor E. Brodsky of the University of Pennsylvania, identified a "back-up alarm" system in host cells that responds to a pathogen's attempt to subvert the immune system.
"In the context of an infection, the cells that are dying are talking to the other cells that aren't infected," said Brodsky, an assistant professor in the Department of Pathobiology in Penn's School of Veterinary Medicine and senior author on the study. "I don't think of it as altruistic, exactly, but it's a way for the cells that can't respond any longer to still alert their neighbors that a pathogen is present."
The findings address the long-standing question of how a host can generate an immune response to something that is designed to shut off that very response. A potential future application of this new understanding may enable the cell-death pathway triggered by bacteria to be harnessed in order to target tumor cells and encourage their demise.
The work appears in the Journal of Experimental Medicine.
A major way that the immune system recognizes pathogens is by detecting patterns that are shared among microbes but are distinct from a host's own cells. Pathogens, however, don't make it easy for immune cells to destroy them. Some can inject proteins into host cells that interfere with this detection, allowing an infection to become established.
Yersinia bacteria, certain species of which cause plague and gastrointestinal disease in humans, is one such pathogen. These bacteria inject a protein, YopJ, into immune cells, that interferes with key signaling pathways, thereby blocking the production of cytokines that could otherwise communicate with other cells about the infection and inducing apoptosis, a form of cell death, which had generally been thought to be non-inflammatory — in other words, a quiet death.
Yet humans and mice can survive Yersinia infections because somehow their immune systems become aware of the presence of an invader.
To understand how host cells overcome Yersinia's insidious strategy, Brodsky's team focused on the activity of an enzyme called RIPK1. RIPK1 was known to play a key role in signaling in respose to an immune cell detecting pathogen-associated patterns as well as inducing cell death.
"RIPK1 sits at a key decision point for the cell," Brodsky said. "Depending on the stimuli the cells see, this protein can transduce a signal to activate gene expression, programmed cell death, or apoptosis, or it can activate another form of cell death called programmed necrosis."
Two recent papers in Nature Cell Biology describe the mechanism of how RIPK1 helps a cell switch between pro-survival and pro-death functions. And while it was known that interfering with this pathway can induce cells to die, there had never been a good physiological explanation for why that should be the case.
The resarchers relied upon a strain of mouse, created by GlaxoSmithKline, that possesses a specific mutation in RIPK1 that renders the enzyme unable to trigger the apoptosis pathway upon encountering Yersinia bacteria.
"This mouse was really useful for us to be able to distinguish between the inflammatory response and apoptosis," Brodsky said.
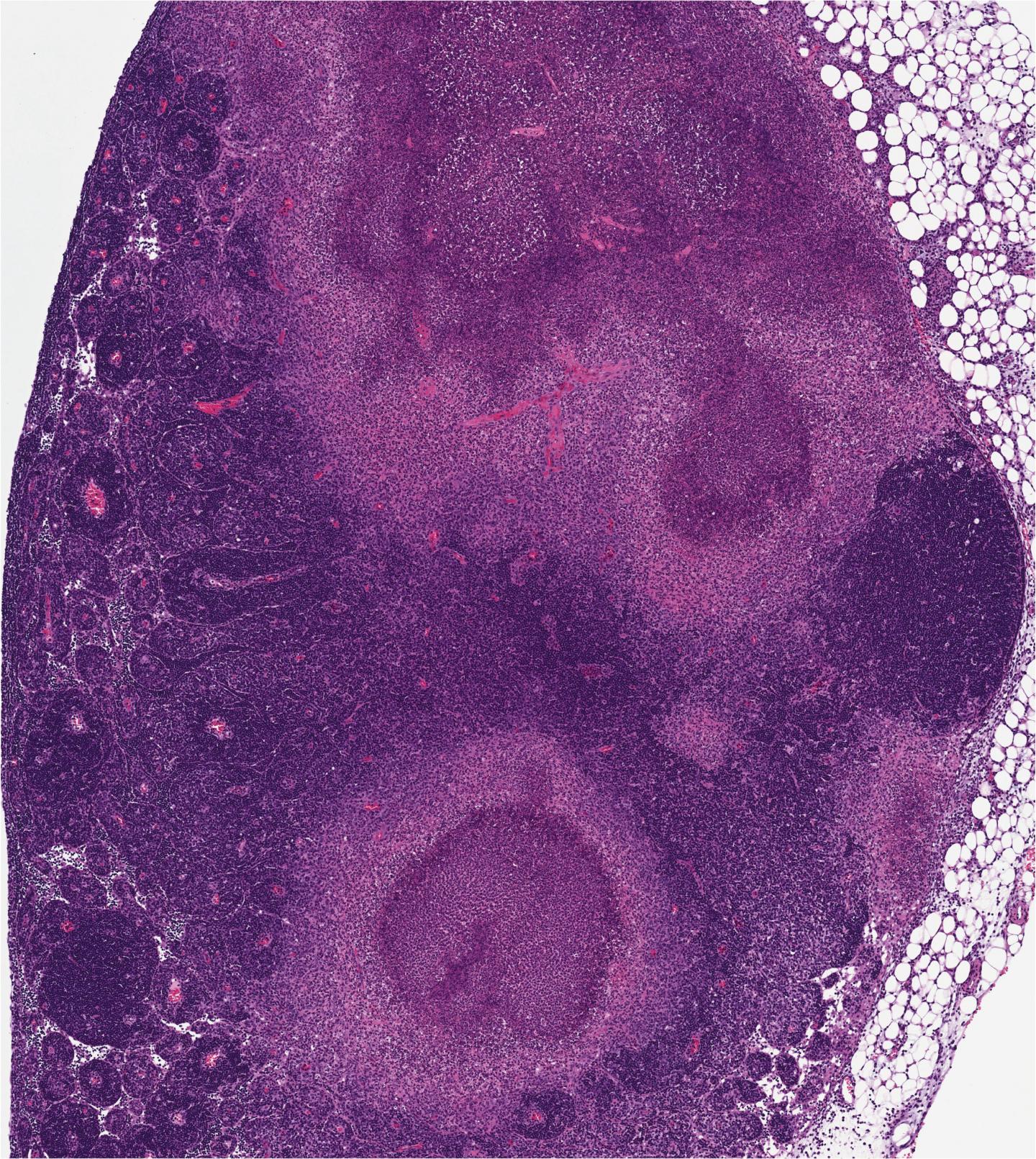
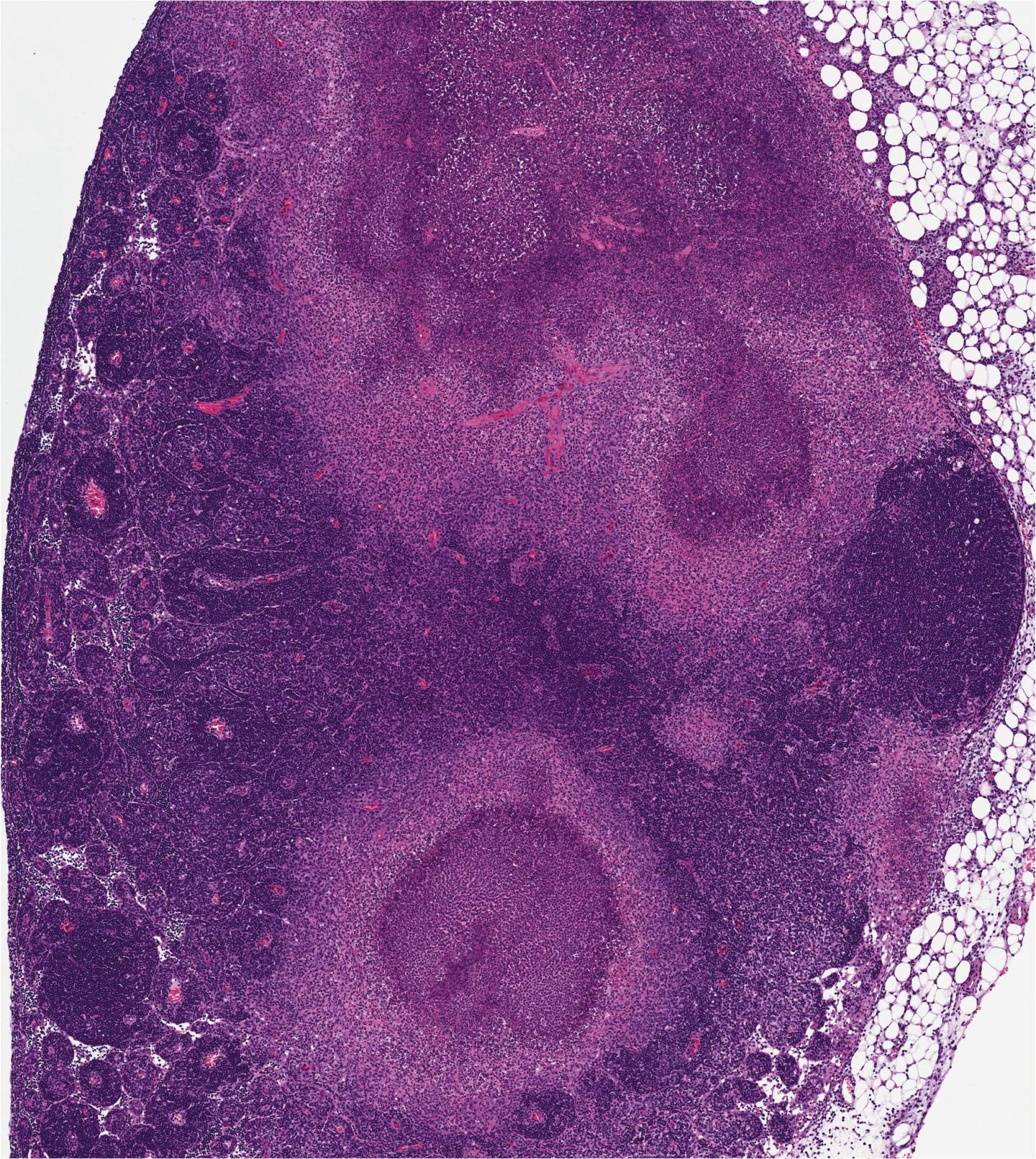
When these mice were infected with Yersinia, their cells did not undergo apoptosis. Instead, these animals became extremely sensitive to infection, succumbing to an infection that normal mice almost always survive. Bacteria could be found dispersed throughout the body, whereas in normal mice Yersinia was typically confined to the lymph nodes, spleen and liver.
"They were unable to control the bacterial burden," Brosky said. "The bacteria disseminated systemically, including to the lungs."
While apoptosis is normally considered non-inflammatory, the researchers showed that RIPK1-induced apoptosis itself promotes cytokine production, presumably by uninfected bystander cells, which helps in recruiting an inflammatory response and plays a role in promoting survival of the host.
The researchers also noticed that the infected lymph nodes from mice that could not undergo RIPK1-induced apoptosis were unable to form granulomas, areas of organized immune cells that can form in response to many infectious or inflammatory stimuli, and are thought to be places of bacterial containment. This suggested that RIPK1-induced apoptosis might promote containment of the bacteria by allowing the immune system to form these granulomas.
Though Brodsky said the research is still at an early stage, one possible therapeutic implication of the work could serve as a way to push cancer cells, which typically grow and thrive without hindrance from the immune system, to their own death.
"We could imagine that modifying bacteria that trigger these pathways, or delivering this bacterial protein to tumor cells, could be potentially useful as an anti-cancer therapeutic," he said.
In future work, Brodsky and colleagues will further investigate the signals that infected cells release in order to start cytokine production by bystander cells. They hope to decipher which molecular pathways are most important in the process. The researchers would also like to develop a more detailed understanding of how granulomas form.
###
Brodsky's coauthors were Penn Ph.D. students Lance W. Peterson, Naomi H. Philip, Alexandra DeLaney, Ruth Choa and Elisabet Bjanes; Penn Vet's Meghan A. Wynosky-Dolfi, Kendra Asklof, Falon Gray, Elisabeth L. Buza and Baofeng Hu; St. Jude Children's Research Hospital's Christopher P. Dillon and Douglas R. Green; and GlaxoSmithKline's Scott B. Berger, Peter J. Gough and John Bertin.
The study was supported by the National Institutes of Health (grants AI103062, AI128530 and AI109267), Burroughs Wellcome Fund, Training Program in Rheumatic Diseases and National Science Foundation.
Media Contact
Katherine Unger Baillie
[email protected]
215-898-9194
@Penn
http://www.upenn.edu/pennnews
Related Journal Article
http://dx.doi.org/10.1084/jem.20170347



{kind=link}