@Blagosklonny concluded in his Aging-US Research Output that here he discussed new evidence that normal aging is not caused by accumulation of molecular damage or telomere shortening
Credit: Correspondence to: Mikhail V. Blagosklonny email: [email protected]
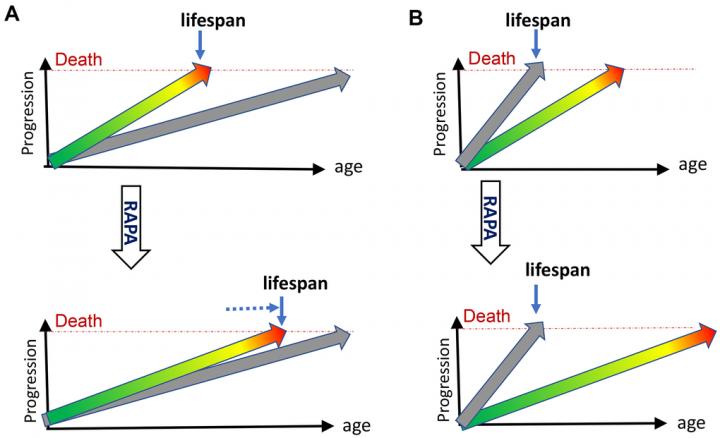
Aging-US published “DNA- and telomere-damage does not limit lifespan: evidence from rapamycin” which reported that failure of rapamycin to extend lifespan in DNA repair mutant and telomerase-knockout mice, while extending lifespan in normal mice, indicates that neither DNA damage nor telomere shortening limits normal lifespan or causes normal aging.
Dr. Mikhail V. Blagosklonny said, “As a provocative title has recently announced, ‘rapamycin fails to extend lifespan in DNA repair -deficient mice’ [1]. The word ‘fails’ implies bad news. Rapamycin tried but failed. Yet, it is expected that the anti-aging drug rapamycin should not restore lifespan of short-lived mice that fail to grow and die young from causes other than normal aging [2]. In such growth- retarded mice, rapamycin, an inhibitor of cell growth, further retards weight gain.”
While shortening lifespan by 18% in unnatural telomerase- deficient mice, in the same study in natural mice, rapamycin increased lifespan by 39% and healthspan by 58%.
In dozens of independent studies, rapamycin has not failed to extend lifespan in normal mice.
However, while extending lifespan in normal mice, rapamycin may fail to save animals dying young from cellular growth retardation.
The failure of rapamycin to extend lifespan in these short- lived mice, dying from DNA damage, rules out the damage theory of aging and to illustrate this point the author first discusses what limits animal lifespan by providing commentary on 1. Quasi-programmed (hyperfunctional) aging and 2. How molecular damage can become life- limiting
Blagosklonny concluded in his Aging-US Research Output that here he discussed new evidence that normal aging is not caused by accumulation of molecular damage or telomere shortening: while extending normal lifespan in mice, rapamycin failed to do so in mice dying from molecular damage.
Previously, several lines of evidence suggested that molecular damage does not cause normal aging. Their detailed discussion is beyond the focus of this article, so he just mentions some of them, without referencing them.
1. Overexpression of enzymes that decrease damage does not extend lifespan in most studies. Similarly, antioxidants do not extend lifespan in animals and may increase mortality in humans. Furthermore, even data that support damage theory can be explained by other mechanisms. For example, N-Acetyl-L-Cysteine, a commonly used anti- oxidant, can inhibit mTOR.
2. According to calculations, molecular damage, especially mtDNA mutations and telomere shortening, cannot reach a deadly threshold during animal lifetime.
3. Genetic knockout of signaling pathways can extend lifespan without affecting molecular damage. Similarly, pharmacological interventions can extend life without affecting damage accumulation.
4. Dramatic intra- and inter-species differences in lifespan poorly correlate with the rate of molecular damage.
5. Nuclear transfer and nuclear reprogramming both rule out DNA damage as a cause of aging. Following adult somatic cell nuclear transfer, cloned animals are healthy and have normal lifespan.
6. Low levels of molecular damage may increase longevity. This phenomenon is known as hormesis. Regardless of mechanistic explanations, this indicates that molecular damage is not-life-limiting even when moderately increased.
7. Rapamycin increases lifespan in all normal animals tested, indicating that mTORC1-dependent quasi-program is life-limiting.
Once again, damage accumulates and must cause death eventually, but quasi-programmed aging terminates life first. Molecular damage can become life-limiting, when artificially accelerated or, potentially, when quasi-programmed aging is decelerated.
###
Sign up for free Altmetric alerts about this article
DOI – https:/
Full Text – https:/
Correspondence to: Mikhail V. Blagosklonny email: [email protected]
Keywords: quasi-programmed aging, hyperfunction theory, antagonistic pleiotropy, natural selection, mTOR
About Aging-US
Launched in 2009, Aging-US publishes papers of general interest and biological significance in all fields of aging research as well as topics beyond traditional gerontology, including, but not limited to, cellular and molecular biology, human age-related diseases, pathology in model organisms, cancer, signal transduction pathways (e.g., p53, sirtuins, and PI-3K/AKT/mTOR among others), and approaches to modulating these signaling pathways.
To learn more about Aging-US, please visit http://www.
Aging-US is published by Impact Journals, LLC please visit http://www.
Media Contact
18009220957×105
[email protected]
Media Contact
Ryan James Jessup
[email protected]
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}