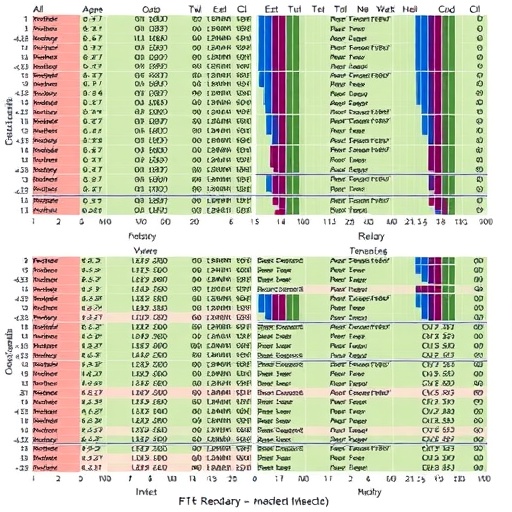
In the rapidly evolving landscape of genomics, the identification of structural variants (SVs) stands at the forefront of breakthroughs that deepen our understanding of evolution, genetic diseases, and gene regulation. A new milestone has been reached with Sniffles2, an advanced, open-source bioinformatics tool designed for precise and comprehensive detection of SVs in long-read sequencing data. This technology is setting unprecedented standards by rapidly and reliably identifying SVs ranging from 50 base pairs to complex genomic rearrangements such as deletions, duplications, insertions, inversions, and translocations.
Structural variants are critical yet challenging to detect mutations that shape genomic diversity. Despite representing the most prevalent form of nucleotide alteration compared to single nucleotide polymorphisms and small insertions/deletions, their detection historically lagged behind due to technical limitations. Sniffles2 emerges as a cutting-edge solution addressing these challenges with superior sensitivity and specificity, leveraging the enhanced resolution afforded by long-read sequencing technologies like those provided by Oxford Nanopore and PacBio.
One of the groundbreaking capabilities of Sniffles2 lies in its reliable detection of mosaic structural variants — SVs present at low variant allele fractions ranging between 5% and 22%. These mosaic variants are particularly elusive but hold significant consequences in oncology and developmental biology, where subpopulations of cells within tissues harbor distinct mutations. The ability to sensitively map these low-frequency variants opens new vistas for research into tumor heterogeneity and somatic mutation-driven diseases.
In addition to single-sample analysis, Sniffles2 excels by offering a joint calling function that integrates multiple samples simultaneously. This feature facilitates comprehensive comparisons such as tumor/normal pair analysis or family trio examinations, enabling researchers to dissect inherited versus de novo variant origins. Moreover, due to its scalable architecture, this tool is well-suited for population-level studies, providing robust structural variant landscapes at unprecedented scale and resolution.
Performance benchmarks position Sniffles2 as a leader in SV calling tools not only in terms of precision but also in computational efficiency. It boasts the ability to process a 40× coverage human genome in approximately 34 CPU minutes or about 8.5 minutes in real-time with standard four-thread execution. This speed gain empowers researchers to perform high-throughput variant discovery within practical time frames that were previously infeasible.
The software is built with user accessibility in mind, aiming to democratize structural variant analysis. By adopting Sniffles2, researchers with foundational expertise in Linux and command-line environments can seamlessly incorporate robust SV discovery into their workflows without needing specialized bioinformatics training. This ease of use heralds a wider adoption of long-read based SV analysis in diverse fields spanning evolutionary biology, medical genetics, and cancer research.
Sniffles2’s methodological framework integrates sophisticated algorithms to parse noisy long-read sequencing data, filtering through background signals to accurately delineate structural variant breakpoints. It applies dynamic clustering of candidate SV signals followed by nuanced consensus building across read evidence, thereby enhancing both sensitivity for true positives and filtering of artifactual calls.
Beyond basic SV identification, the tool supports detailed annotation of detected variants enabling downstream integration with genomic databases and variant interpretation pipelines. These annotations are vital for translating raw calls into biologically meaningful insights, linking structural changes to potential phenotypic consequences or disease mechanisms.
The implications of Sniffles2 extend deeply into clinical contexts, where uncovering causative mutations can revolutionize diagnostic and therapeutic strategies. The software has already demonstrated utility in pinpointing crucial mutations underlying inherited diseases and pivotal alleles driving cancer progression, underscoring its transformative potential in precision medicine.
While short-read sequencing revolutionized variant detection, its limitation in spanning complex genomic regions has left a gap in SV detection. Sniffles2 leverages long-read platforms’ extended read lengths to overcome these barriers, reliably detecting variants that short reads typically miss due to ambiguous alignments or low mappability.
Critical to current and future genomics projects is the seamless integration of tools like Sniffles2 into broader bioinformatics ecosystems. Its compatibility and open-source nature encourage iterative improvements and community-driven enhancements, propelling the field towards more comprehensive and reliable genomic variation maps.
The protocol introduced alongside Sniffles2 offers a detailed, step-by-step approach to implementing the software across different genomic contexts — from standard germline variant calling to sophisticated mosaicism analyses and population-scale joint calls. This guidance ensures both novice and expert users can maximize the tool’s capabilities in their specific research endeavors.
Looking ahead, the adoption of Sniffles2 promises to accelerate discoveries by providing a high-resolution view of the structural genome. This advancement addresses pressing challenges in genomics research, ultimately translating into enhanced understanding of disease etiology, evolutionary processes, and the complex regulatory genome architecture.
In summary, the development and dissemination of Sniffles2 epitomize the gene-centric revolution in genomics, turning long-read sequencing data into actionable knowledge with speed, precision, and accessibility. As the field harnesses this technology, the era of comprehensive structural variant analysis is poised to unlock new frontiers in biology and medicine.
Subject of Research: Structural variant detection using long-read sequencing and bioinformatics methodologies
Article Title: Structural variant calling using Sniffles2
Article References:
Paulin, L.F., Romanek, H., Jaryani, F. et al. Structural variant calling using Sniffles2. Nat Protoc (2026). https://doi.org/10.1038/s41596-026-01362-w
Image Credits: AI Generated
DOI: https://doi.org/10.1038/s41596-026-01362-w
Tags: deletions duplications insertions inversionsevolution and gene regulation genomicsgenetic disease variant analysisgenomic rearrangements identificationlong-read sequencing structural variantslow allele fraction mosaic variantsmosaic structural variant detectionOxford Nanopore SV analysisPacBio sequencing structural variantssensitive and specific SV detectionSniffles2 bioinformatics toolstructural variant detection in genomics
{kind=link}