Drug discovery researchers from El Paso to Galveston join forces to find new therapeutics to combat the pandemic
Credit: Drug Discovery Consortium
As the world prays for the quick discovery of a cure for COVID-19, an international team of computational scientists, medicinal chemists, biochemists, and virologists — led by Texas researchers — have coalesced to rapidly identify drug-like molecules that inhibit SARS-CoV-19 replication.
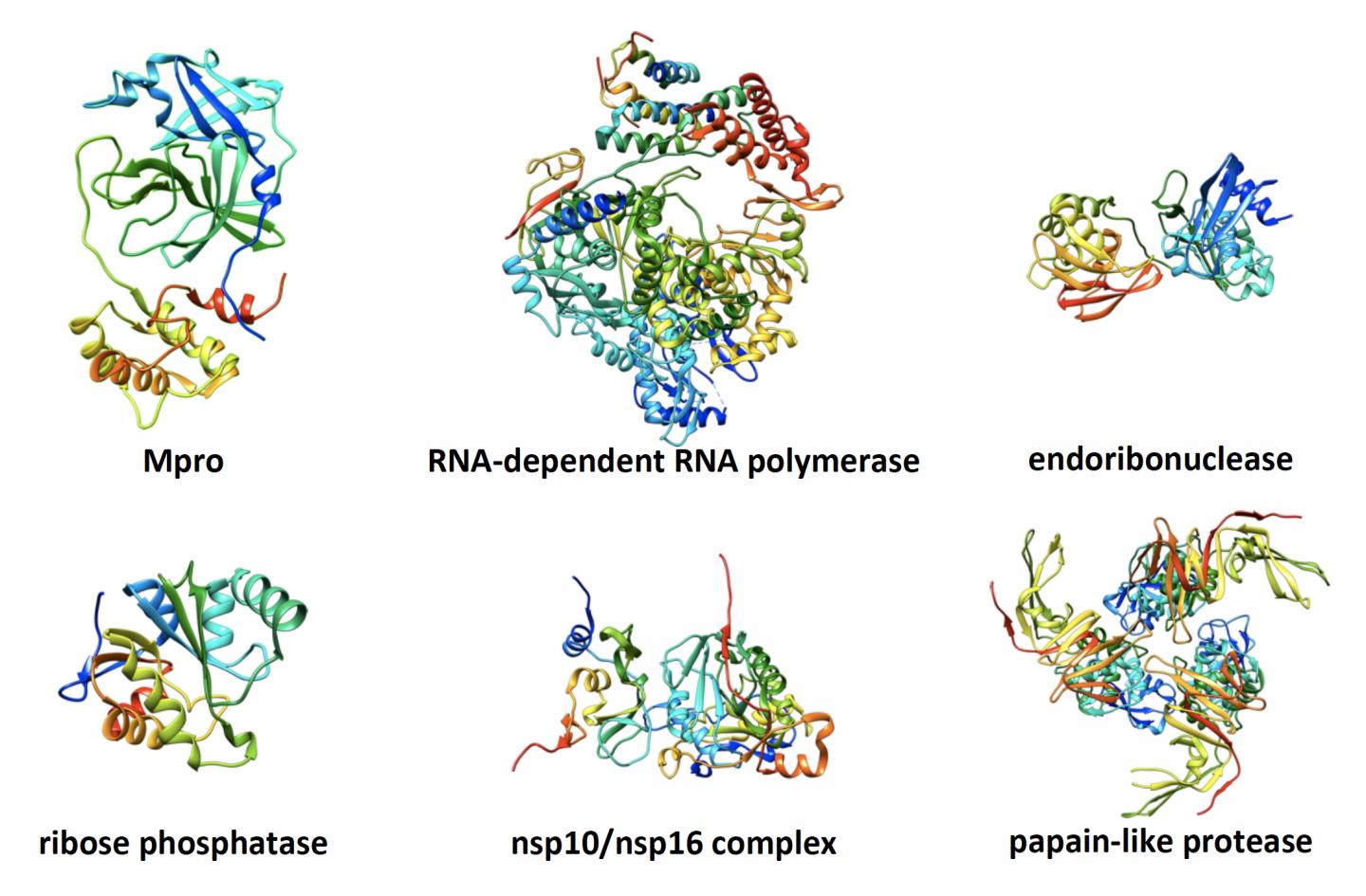
The COVID-19 Drug Discovery Consortium is leveraging some of the world’s most powerful supercomputers to screen millions of small molecules against all the major non-structural proteins of SARS-CoV-19 in order to find molecules that stop the virus.
Based on the results of their computational drug screening, they are moving quickly to test the most promising drug-like molecules against the virus in high containment facilities. The most potent antiviral compounds will transition to manufacturing and safety/toxicity studies in preparation for first-in-human clinical studies.
The effort grew out of a group of researchers who had previously used the Drug Discovery @ TACC portal — a collaboration between Stan Watowich and Usha Viswanathan at the University of Texas Medical Branch (UTMB) and developers at the Texas Advanced Computing Center (TACC).
“The drug discovery portal lets researchers start with the biological target that is important to them and computationally test out millions of potential drug candidates using the physics of molecular interactions to find the best ones,” said John Fonner, a research associate In the TACC Life Sciences Computing group and one of the portal developers. “From there, researchers pick which candidates to further optimize and test experimentally.”
First launched in 2015 (and building on earlier projects created in the wake of bioterrorism scares stretching back to 2005), the portal has helped hundreds of researchers find promising drug leads for other, non-coronavirus, diseases and has supported millions of hours of computational work. Refocusing on COVID-19 was a natural fit.
“We uniquely positioned for this,” Watowich asked. “We’ve done it for a long time. We know lots of players. And we might be able to make a difference quickly. There won’t be a drug in the next month, but we can identify a number of new avenues that can be developed into a drug in the next 10 months.”
BUILDING THE CONSORTIUM
Starting in March 2020, Watowich put out a call to users of the portal to organize a communal response to COVID-19 and determine what was needed to advance the cause.
“It would be a waste of time to reproduce research,” he said. “So instead, we developed a consortium to minimize overlap and focus on getting something done quickly.”
The consortium’s more than 30 researchers settled on seven potential viral protein targets to screen drug-like molecules against — parts of SARS-CoV-2 that the group believed had vulnerabilities that a drug could exploit.
For each of the proteins, there were several potential structures, or 3D models of the virus that show where a drug might bind in such a way as to change the virus’s shape or disrupt its function. The team came up with 20 likely structures with which the drug docking molecules would virtually interact.
By community consensus, the consortium focused on immediately testing three libraries of commercially-available small molecules for possible efficacy against their SARS-CoV-2 targets.
These libraries contain 2.6 million digital analogues of small molecule chemicals that are readily available for purchase from Enamine Chemical Company, the world’s largest provider of screening compounds, building blocks, and fragments. The advantage of working with Enamine is that any computer “hits” — promising molecules that seem to disrupt COVID-19 — can be purchased and tested immediately.
Rather than working independently, the consortium coordinated their efforts: tabulating what targets people were looking on, what library they screened, what conditions they used, and what the results were.
Suman Sirimulla, an assistant professor of Pharmacy at UT El Paso who set up the docking calculations, served as the informal gatekeeper, engaging the community and making sure researchers did not duplicate work.
Once their path was set, the team used the Stampede2 and Lonestar5 supercomputers at TACC to rapidly churn through calculations, completing roughly 50 million binding calculations a day using hundreds of TACC supercomputer cores in parallel.
“The studies we’re trying to do are not feasible on small compute clusters,” Sirimulla said. “Screening millions of compounds against many proteins — that’s a lot of computation and we want to get this done in a few days. So we need clusters like TACC’s.”
By May 6, the consortium’s computations on Lonestar5 and Stampede2 identified the top 600 candidates in the libraries — “a reasonable number to screen in high-level biocontainment facilities,” Watowich explained. “The robots that are available in the biocontainment lab can screen hundreds of compounds at a time, so we should know how our candidates perform against SARS-CoV-2 in a matter of weeks.”
TESTING, TESTING, TESTING
The drug screening computations are only the starting point. Through contacts at Boston University, Texas A&M University, UTMB, and Enamine, Watowich received commitments to physically make a library of the compounds that their calculations predicted would be effective and have these compounds tested to see if they work and are safe to use.
“The calculations are there to provide a prediction; and a prediction is useless if you don’t have the means and passion to test it,” Watowich said. “I wasn’t going to get involved in this effort if we couldn’t generate predictions that we can actually test.”
The compounds will be tested in the level 4 screening facility at Boston University, one of only about handful in the nation that can study live SARS-CoV-2 viruses.
Concurrently, researchers at the Texas A&M Institute of Biosciences and Technology in Houston will profile the library of compounds for activity in cell cultures, enabling researchers to focus their attention on compounds showing the greatest selectivity in anti-viral activity. Based on these preliminary findings, researchers at UTMB’s level 3 screening facility, one of only two national laboratories in the nation dedicated to combating infectious threats to health, will perform follow-up tests on the most active and selective test molecules.
“One month from now, we’ll know if we found new drug-like molecules that haven’t been looked at before that pharmaceutical companies can make modifications to and produce quickly,” Watowich said.
Some might ask, given the high profile testing of drugs like remdesivir, and the global effort to design a vaccine, whether we need more drug screening studies like the consortium’s?
“Hopefully we don’t,” Watowich said. “But going ahead and saying we won’t need anything, don’t bother, that the drugs under investigation solve the problem and the virus won’t mutate, is a mistake. In the next year, we may need prophylactics, or drugs that might work for those in nursing homes. There’s lots of reasons why we might need additional compounds. And we don’t know if any of the drugs that are being touted as effective actually are, or work for all people.”
From his own decades-long drug discovery research, Watowich knows that high-throughput computer drug screening can be a shot in the dark, but one that returns results.
“We’ve done this before with other targets in our labs,” he said. “There are lots of cases where screening against libraries gives a starting point. In fact, the company I started is based on this approach.”
Nonetheless, on a good day, five percent of predictions pan out. On a bad day, one percent do.
“It’s a bold, rapid, high-risk, high-reward, initiative to find new starting points for drugs for COVID-19,” he said. “It may strike gold or it may go nowhere. There’s only one way to find out: to do the work and run the experiments.”
###
Media Contact
AARON DUBROW
[email protected]
Original Source
https:/


{kind=link}