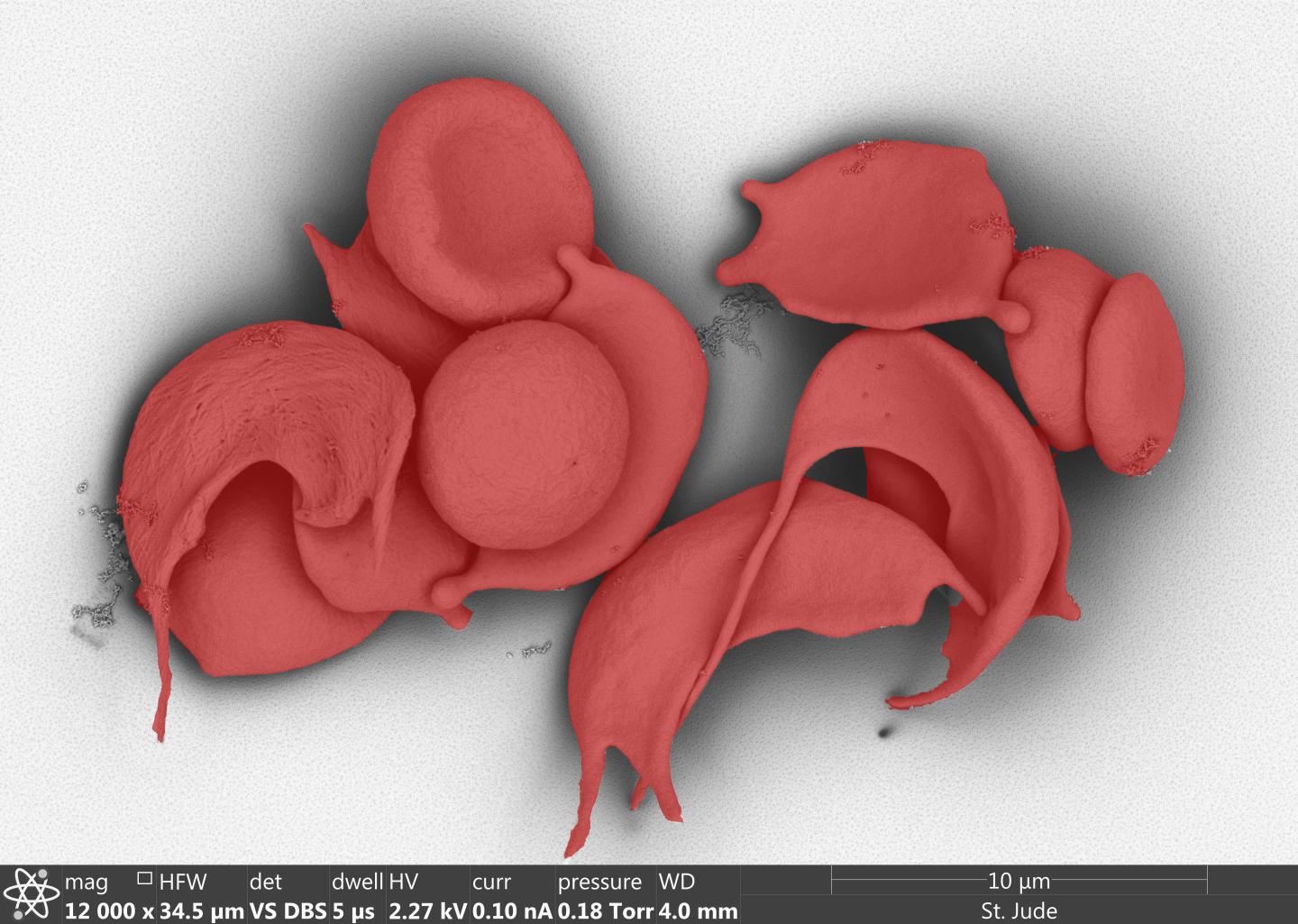
The 2018 annual meeting of the American Society of Hematology will include research from St. Jude Children’s Research Hospital that stretches from the laboratory to the clinic

Credit: Image by Julie Justice; Randall Wakefield; Jennifer Peters, PhD; Christophe Lechauve, PhD; Victoria Frohlich, PhD.
The 60th Annual Meeting of the American Society of Hematology will feature research from St. Jude Children’s Research Hospital on topics ranging from the genomic basis and vulnerabilities of leukemia to an update on gene therapy for hemophilia B to advances in sickle cell disease and beta-thalassemia.
St. Jude research will be highlighted at the Sunday plenary session and throughout the four-day meeting that begins Saturday in San Diego. St. Jude faculty and staff will also chair sessions, give talks, provide updates on clinical advances, participate in panels and moderate sessions on scientific and clinical topics throughout the meeting and in the Friday scientific workshops. The subjects include inherited predisposition to hematopoietic malignancies, how to establish a hematopoietic predisposition clinic and myeloid development.
At the plenary session, St. Jude researchers will lift the veil on the biology of acute erythroid leukemia (AEL) to reveal the genomic basis and therapeutic vulnerabilities of this high-risk leukemia. The findings will be presented by Ilaria Iacobucci, Ph.D., a scientist in the laboratory of Charles Mullighan, MBBS, M.D., a member of the St. Jude Department of Pathology. The plenary session begins at 2 p.m. PT in San Diego Convention Center Hall AB.
“This project marks a paradigm shift in our understanding and possible treatment of acute erythroid leukemia,” Mullighan said. AEL is a rare subtype of acute myeloid leukemia (AML) that affects children and adults. AEL has a poor prognosis. Until this study, the genetic basis was poorly understood.
Researchers previously used integrated genomic analysis to compare 159 patients with AEL to 1,509 patients with AML or related red-blood disorders. Scientists identified five age-related subtypes of AEL, each with distinct genomic features and survival rates.
Using CRISPR/Cas9 genome editing, researchers developed mouse models of the AEL subtypes. This was done by simultaneously inactivating different combinations of nine genes in mouse blood stem and progenitor cells. The genes were frequently mutated in the AEL subtypes. The method helped to capture the genomic complexity of AEL and identify mutations driving the leukemia. For example, inactivating the genes Tp53, Bcor and Dnmt3a or Tp53, Bcor, Rb1 and Nfix promoted development of AEL. Inactivating a different constellation of genes–Tp53, Bcor, Tet2–promoted development of B-ALL.
The mouse models were then used to screen the subtypes for sensitivity to almost 200 chemotherapy agents and other compounds, including precision medicines that target specific pathways.
“We found that leukemic cells driven by different mutations have different sensitivities and vulnerabilities to drugs,” Iacobucci said. For example, AEL subtypes with NUP98-fusions were highly sensitive to various classes of BET inhibitors and were resistant to common chemotherapeutic drugs. Tp53 wild-type tumors without gene fusions were highly sensitive to a variety of standard chemotherapy agents. In contrast, leukemia subtypes with Tp53 mutations resisted certain chemotherapy drugs, but were sensitive to compounds called PARP inhibitors.
Mullighan said: “These results make a strong case for using genomics to classify patients and then use the information to help guide therapies, since leukemic cells with different genotypes do not respond to treatment the same way.”
Other St. Jude oral presentations:
Sunday
San Diego Convention Center, Room 25B
Abstract: 385
Noon PT
Leukemia risk gene revealed as an important regulator of B-cell development
Certain genetic variation in the gene ARID5B is linked to as much as a two-fold increased risk of childhood acute lymphoblastic leukemia (ALL), but until now the biology behind this association and the function of the ARID5B protein have been poorly understood.
Charnise Goodings-Harris, Ph.D., a St. Jude postdoctoral fellow in the laboratory of Jun J. Yang, Ph.D., an associate member of the Department of Pharmaceutical Sciences, will present evidence Sunday that ARID5B is an important regulator of B-cell development. “These results fill a knowledge gap regarding the roles this protein plays during blood cell development,” Yang said. “That can then help us understand how variations in the gene influence risk of pediatric ALL.”
For this study, researchers developed a mouse that overexpressed Arid5b and tracked how that affected development of B cells and other blood components. B-ALL is the most common childhood cancer.
Previous work by Yang and others identified inherited variations in ARID5B that were associated with elevated susceptibility to ALL in childhood and to a poor prognosis. Compared to children of European ancestry, children of Hispanic ethnicity had more of the high-risk variations and had an increased risk of developing the disease.
San Diego Convention Center, Room 25B
Abstract: 410
4:45 p.m. PT
Researchers home in on the mechanism to turn back the clock on hemoglobin production
Sunday, St. Jude researchers will offer new details of the mechanism by which red blood cells switch from producing fetal hemoglobin to adult hemoglobin. That change leaves individuals with the inherited blood disorders sickle cell disease and beta-thalassemia at risk for debilitating, sometimes life-threatening complications. Increasing fetal hemoglobin levels is proven to ease symptoms in affected individuals.
For this study, researchers identified the protein UHRF1 in a gene editing-based screen designed to find proteins that facilitate the fetal-to-adult hemoglobin switch.
“UHRF1 is the guardian of methylation throughout the genome,” said senior author Mitchell Weiss, M.D., Ph.D., chair of the St. Jude Department of Hematology. Methylation, the process for adding or removing methyl groups from DNA, regulates gene expression.
“In this study, we detail efforts to pinpoint where methylation is most important for turning off fetal hemoglobin with the goal of reactivating it in individuals with sickle cell disease or beta-thalassemia to alleviate their symptoms. This study contributes to that effort,” Weiss said.
Ruopeng Feng, Ph.D., a postdoctoral fellow in the Weiss laboratory, will present the results.
San Diego Convention Center, Room 25B
Abstract: 411
5 p.m. PT
Immunosuppression agent may offer new treatment of beta-thalassemia
An immunosuppressive drug best known for protecting organ transplants from rejection may also help to ease symptoms of beta-thalassemia, a common inherited blood disorder. The research will be presented Sunday by Christophe Lechauve, Ph.D., scientist in the laboratory of Mitchell Weiss, M.D., Ph.D., chair of the St. Jude Department of Hematology.
The drug is rapamycin. St. Jude researchers found that rapamycin reduced the effects of beta-thalassemia in mice and in cultured red blood cells of beta-thalassemia patients. Rapamycin combats -thalassemia by activating an enzyme (Ulk1) that reduces the toxic build-up of hemoglobin components.
Hemoglobin is the protein in red blood cells that carries oxygen. Normal hemoglobin has four protein chains–two alpha-globin and two beta-globin. Beta-thalassemia patients have too little beta-globin, which leads to a toxic build-up of ?-globin in red blood cells and the developing cells that give rise to them. Patients develop anemia and other, sometimes life-threatening, symptoms associated with the disorder.
“Rapamycin treatment significantly reduced and in some cases fully corrected the accumulation of ?-globin in the developing blood cells of patients with beta-thalassemia,” Weiss said. Researchers hope the findings will lead to a clinical trial of the drug in beta-thalassemia patients.
San Diego Convention Center, Room 6B
Abstract: 491
5:30 p.m. PT
Gene therapy provides long-term relief for men with severe hemophilia
Eight years after the first men with severe hemophilia B received gene therapy developed in Memphis and London, researchers will report the therapy is still working. The therapy was pioneered by researchers at St. Jude, University College London and the Royal Free Hospital.
“This is the first report that gene therapy provides a safe, reliable supply of the blood clotting factor for such an extended period after a single infusion,” said co-author Ulrike Reiss, M.D., of the St. Jude Department of Hematology. “The findings address lingering concerns that factor IX levels might decline with time.”
Not only were the initial increased levels in clotting factor IX maintained in the 10 men enrolled in the study, but their bleeding episodes decreased 82 percent. Their use of clotting-factor concentrate to prevent or treat spontaneous bleeds dropped 66 percent.
A mutation in the factor IX gene leaves individuals with hemophilia B, who are mostly men, with sometimes dramatically reduced or absent levels of the clotting factor. That leaves patients at risk for painful episodes of spontaneous bleeding that can result in crippling joint damage early in life as well as potentially fatal bleeds in the brain.
For this study researchers modified the adeno-associated virus (AAV8) to serve as the delivery device (vector) to carry the genetic material for making factor IX in liver cells, where it is normally produced.
Researchers also reported that a new vector preparation method failed to prevent an asymptomatic increase in liver enzymes that some patients experienced. Enzyme levels returned to the normal range following steroid treatment. Investigators had tried removing empty viral particles before the gene therapy was infused into patients in case they were triggering an immune response. This finding suggests other factors are involved.
While this study is closed to new patients, St. Jude, University College London and the Royal Free Hospital have opened another gene therapy trial for patients with severe hemophilia B. This clinical trial involves a new vector that researchers believe is even more effective.
Monday
Marriott Marquis San Diego Marina, Pacific Ballroom 24
Abstract: 529
7 a.m. PT
Tumor suppressor protein also plays key role in red blood cell development
St. Jude researchers have identified an intriguing new role for the tumor suppressor protein FBXO11 and a new mechanism important for red blood cell maturation.
FBXO11 is a type of enzyme called ubiquitin ligase, which tags unneeded or unwanted proteins for degradation. Previous studies showed that loss-of-function mutations in the FBOX11 gene arrest the development of normal lymphoid cells and promote their transformation to cancer. In this study, researchers found that FBXO11 is essential for the development of human and mouse red blood cells.
“Evidence suggests that FBXO11 acts early in red blood cell development to degrade a repressor protein called BAHD1, thereby activating the expression of hundreds of genes essential for maturation of red blood cells,” said Mitchell Weiss, M.D., Ph.D., chair of the St. Jude Department of Hematology. “The finding may help explain how FBXO11 works in other tissues and prevents cancer.”
Peng Xu, Ph.D., a postdoctoral fellow in Weiss’ laboratory, will present the research Monday.
San Diego Convention Center, Room 25B
Abstract: 565
7 a.m. PT
RNA sequencing helps uncover mutations that launch most common childhood cancer
Researchers led by St. Jude have identified eight new subtypes of the most common form of acute lymphoblastic leukemia (ALL), which is also the most common childhood cancer. The finding means that more than 90 percent of B-ALL cases can now be classified by subtype.
“Prior to this study, 30 percent of B-ALL cases could not be classified into subtypes,” said corresponding author Charles Mullighan, MBBS, M.D., of the St. Jude Department of Pathology. “These patients lacked precision medicine approaches to treatment and commonly relapsed.”
The research, which identified 23 B-ALL subtypes, involved integrated genomic analysis, including RNA sequencing, of almost 2,000 children and adults with B-ALL. The research highlighted RNA sequencing as a tool to recognize chromosomal rearrangements, gene-expression profiles, novel point mutations and other alterations that are difficult to detect using whole-genome or whole-exome sequencing.
The newly identified subtypes are strongly associated with prognosis in children and adults. The list includes two that involve the PAX5 gene, notably PAX5 P80R, the first point mutation identified that initiates leukemia. Researchers reported that the mutation blocked B cell differentiation and maturation, and in a mouse model created by CRISPR/Cas9 genome editing, resulted in the development of leukemia in mice. The PAX5 subtypes accounted for almost 10 percent of previously uncategorized B-ALL cases.
The study provides a new framework for classifying ALL in children and adults and shows the utility of RNA-sequencing to provide much of the information required for accurate diagnosis of B-ALL.
Zhaohui Gu, PhD, a postdoctoral fellow in Mullighan’s laboratory, will present the results Monday. . St. Patient-derived samples of many B-ALL subtypes are available to researchers worldwide through a St. Jude resource called PROPEL (Public Resource of Patient-derived and Expanded Leukemias). The PROPEL data portal provides access to more than 200 human leukemia samples. The cancers were grown in mice and are known as patient-derived xenografts.
San Diego Convention Center, Room 10
Abstract: 550
7:45 a.m. PT
Gene rearrangement drives high-risk leukemia
Charles Mullighan, MBBS, M.D., of the St. Jude Department of Pathology, and his colleagues recently identified a subtype of acute leukemia defined by rearrangements involving the transcription factor gene ZNF384. These tumor cells typically have features of both B lymphocytes and myeloid cells, and may be diagnosed as B-ALL or mixed phenotype acute leukemia (MPAL).
Researchers had shown that the ZNF384 fusion proteins cause leukemia when expressed in mouse bone marrow, and that the fusions are first acquired in primitive human blood stem cells.
This study examined the effects of expressing the fusion in human blood stem cells. Using stem cells isolated from umbilical cord blood, the researchers showed that the TCF3-ZNF384 fusion, but not normal ZNF384, skewed blood cell development to resemble that of human leukemic cells. In addition, the stem cells resulted in development of B/myeloid leukemia in mice.
“This is the first human model of mixed phenotype leukemia, and provides the foundation to probe the mechanism, including associated epigenetic changes, induced by the fusion gene,” Mullighan said. Patient-derived samples of the subtype are available to researchers through the PROPEL data portal.
Kirsten Dickerson, a graduate student in the Mullighan laboratory, will present the research Monday.
San Diego Convention Center, Room 29C
Abstract: 723
3:15 p.m. PT
Online resource offers rich source of genomic and clinical data on sickle cell disease
St. Jude researchers have developed a data portal to fuel collaboration and advance understanding of the genomic landscape of sickle cell disease, particularly genetic factors that contribute to disease severity and complications. Lance Palmer, Ph.D., of the St. Jude Department of Computational Biology, will outline the initiative during his presentation Monday.
The Sickle Cell Disease Portal includes whole-genome sequencing data and select clinical information from more than 800 individuals with the chronic inherited blood disorder. It is one of the first such sickle cell disease data sets available to researchers worldwide. The resource is expected to grow as whole- genome sequencing data from more individuals with sickle cell disease are added as well as visualization tools for scientists.
Each year about 300,000 infants are born worldwide with sickle cell disease. It is caused by inherited mutations in the HBB gene that results in stiff, brittle red blood cells with a characteristic sickle shape. Affected individuals are at risk for premature death and a variety of complications, including episodes of acute pain, stroke and progressive organ damage.
“Not every patient gets every complication,” said Mitchell Weiss, M.D., Ph.D., chair of the St. Jude Department of Hematology. “Genetics plays a role in who develops which complications, but many of those genetic modifiers are unknown or poorly understood.
“The Sickle Cell Disease Portal is designed to complement existing resources and help address the knowledge gaps by fueling the collaboration that is essential to change the outcomes for patients,” he said.
The portal includes whole-genome sequencing data of 503 St. Jude patients, primarily individuals enrolled in the Sickle Cell Clinical Research and Intervention Program (SCCRIP). This long-term St. Jude study follows individuals with sickle cell disease throughout their lives. The goal is to better understand how the disease progresses and assess treatment efficacy.
The site also includes whole-genome sequencing data from 304 sickle cell patients treated at Baylor College of Medicine in Houston.
Overall, more than 98 percent of the Sickle Cell Disease Portal data come from African-American participants. Along with whole-genome sequencing, the site includes such clinical information as hemoglobin and fetal hemoglobin levels as well as some known genetic modifiers of the disease. Identifying patient information has been removed.
The information is available through St. Jude Cloud, a data-sharing resource developed to make it easier for scientists worldwide to access raw sequencing data along with analytic and visualization tools for pediatric cancer and sickle cell disease.
Data-access requests will be handled by a data-access committee. Along with whole-genome sequencing data, access may include coded clinical and demographic information.
###
St. Jude media relations staff onsite at ASH
Michael Sheffield
Cell: (901) 379-6072
Katy Hobgood
Cell: (901) 568-9869
St. Jude Children’s Research Hospital
St. Jude Children’s Research Hospital is leading the way the world understands, treats and cures childhood cancer and other life-threatening diseases. It is the only National Cancer Institute-designated Comprehensive Cancer Center devoted solely to children. Treatments developed at St. Jude have helped push the overall childhood cancer survival rate from 20 percent to 80 percent since the hospital opened more than 50 years ago. St. Jude freely shares the breakthroughs it makes, and every child saved at St. Jude means doctors and scientists worldwide can use that knowledge to save thousands more children. Families never receive a bill from St. Jude for treatment, travel, housing and food — because all a family should worry about is helping their child live. To learn more, visit stjude.org or follow St. Jude on social media at @stjuderesearch.
Media Contact
Michael Sheffield
[email protected]
901-379-6072
Original Source
http://www.

{kind=link}