In the relentless battle against breast cancer, one of the most formidable obstacles remains endocrine resistance, particularly within hormone receptor–positive (HR+) breast cancer subtypes. Despite advances in targeted therapies, the emergence of resistant tumors significantly undermines treatment success and patient prognosis. Intriguingly, a substantial proportion of HR+ breast cancers—up to 70%—exhibit overexpression of the human epidermal growth factor receptor 3 (HER3), a receptor tyrosine kinase implicated in driving tumor progression and therapeutic evasion. A novel therapeutic agent, HER3-DXd (patritumab deruxtecan), an antibody-drug conjugate currently undergoing clinical evaluation, directly targets HER3-expressing metastatic breast cancers. Yet, the pressing challenge remains: how can this therapeutic be optimized, especially in the context of endocrine-resistant disease?
Recent groundbreaking research suggests a compelling answer lies in the strategic inhibition of ATR (ataxia telangiectasia and Rad3-related protein), a critical kinase orchestrating the cellular DNA damage response (DDR). The DDR is essential for maintaining genomic integrity, particularly under stress conditions such as those induced by cancer therapies. ATR functions as a guardian of genome stability, triggering cell cycle arrest and activating DNA repair pathways in response to replication stress and DNA damage. Recognizing this, researchers hypothesized that pharmacological blockade of ATR could potentiate the antitumor cytotoxicity of HER3-DXd by amplifying DNA damage and curtailing repair mechanisms in HER3-positive/HR-positive breast cancer cells, including models resistant to tamoxifen, a commonly used endocrine therapy.
This hypothesis stems from a mechanistic insight into how cancer cells evade therapy-induced cell death. HER3-DXd operates by delivering cytotoxic payloads directly to HER3-overexpressing cancer cells, causing DNA damage that ideally culminates in apoptosis. However, the cancer cells’ intrinsic DDR machinery often mitigates this assault by repairing DNA lesions, contributing to therapeutic resistance. By interrupting ATR signaling, these repair pathways can be dismantled, potentially overwhelming the tumor cells with unrepaired DNA damage and pushing them toward irreversible cell death.
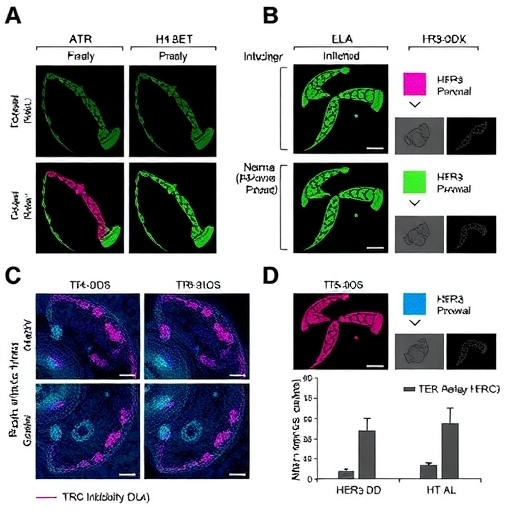
Experimental validation of this approach was conducted using preclinical models representative of HR+ breast cancer, with a focus on tamoxifen-resistant phenotypes. The researchers demonstrated that combining HER3-DXd with ATR inhibitors significantly increased DNA damage markers within tumor cells compared to either treatment alone. These findings indicate a synergistic interaction whereby ATR inhibition amplifies the genotoxic stress induced by HER3-DXd. Consequent analyses revealed heightened levels of DNA double-strand breaks and replication stress, hallmarks of effective anticancer treatment infliction.
Functional assays further confirmed that the dual treatment severely impaired tumor cell viability and clonogenic potential. Importantly, this effect was markedly pronounced in tamoxifen-resistant cell lines, suggesting that ATR inhibition could sensitize tumors that have developed resistance to standard endocrine therapies. This revelation offers a promising avenue for overcoming one of the most vexing therapeutic hurdles in HR+ breast cancer management.
The translational significance of this work cannot be overstated. Despite the clinical promise of HER3-DXd, its efficacy may be limited by the robust DNA repair capabilities in cancer cells. Dual targeting with ATR inhibitors logically extends the therapeutic window, creating a synthetic lethal milieu in which tumor cells succumb due to their inability to rectify lethal DNA damage. Such combination strategies could redefine treatment paradigms, particularly for patients whose tumors have ceased responding to endocrine agents such as tamoxifen.
Beyond cellular assays, in vivo models corroborated enhanced tumor growth suppression with the combined regimen, reinforcing the therapeutic potential and setting the stage for clinical investigation. These preclinical insights advocate for clinical trials that incorporate ATR inhibitors with HER3-targeted antibody-drug conjugates, which could revolutionize the therapeutic landscape for HER3-positive and endocrine-resistant breast cancer cohorts.
From a molecular perspective, this study elucidates how DNA repair pathways interplay with receptor tyrosine kinase signaling and therapeutic susceptibility. By intricately dissecting these relationships, the research contributes vital knowledge to the cancer biology field, highlighting how the vulnerabilities of DNA damage repair can be exploited in conjunction with targeted delivery of cytotoxic agents.
The implications of ATR inhibition extend beyond breast cancer. Given the ubiquity of DNA repair mechanisms in diverse tumor types, the principles unveiled here may catalyze broader oncology applications. Combination therapies that thwart adaptive repair responses while simultaneously administering targeted cytotoxins could become a versatile strategy against various malignancies harboring similar resistance profiles.
While the promise is significant, challenges remain. The potential toxicity of ATR inhibitors, particularly in combination regimens, requires careful evaluation. The balance between maximizing tumor cell kill and minimizing harm to normal proliferating cells forms an intricate therapeutic tightrope. Ongoing research must address optimal dosing, scheduling, and biomarker development to identify patients most likely to benefit from this approach.
Moreover, the heterogeneity of HER3 expression within tumors and the dynamic evolution of resistance mechanisms call for personalized medicine strategies. Integrating genomic and proteomic analyses may enable precise stratification and monitoring, ensuring that patients receive tailored interventions, enhancing efficacy while curbing adverse effects.
In conclusion, targeting the ATR-mediated DNA damage response pathway represents a compelling advancement in the fight against HER3-positive, hormone receptor–positive breast cancer, particularly in the challenging context of endocrine resistance. The synergism observed with HER3-DXd posits a novel combinational therapy that undermines cancer cells’ repair defenses, accentuating DNA damage and fostering robust antitumor effects. These findings, published in the British Journal of Cancer, open new horizons for translational research and clinical innovation, fueling hope for improved outcomes in metastatic breast cancer patients who currently face limited options.
As with all pioneering treatments, further clinical validation is essential to confirm safety, efficacy, and long-term benefits. Nonetheless, the strategic blockade of ATR to enhance HER3-directed therapeutics may soon herald a new era, promising to surmount one of the most persistent barriers in breast cancer treatment and offering renewed optimism for countless patients worldwide.
Subject of Research:
Targeting ATR to enhance the efficacy of HER3-DXd in hormone receptor–positive, HER3-expressing breast cancer, particularly addressing endocrine therapy resistance.
Article Title:
ATR inhibition potentiates the antitumor efficacy of HER3-DXd in HER3-positive/HR-positive breast cancer by increasing DNA damage.
Article References:
Xie, X., Lee, J., Gi, Y.J. et al. ATR inhibition potentiates the antitumor efficacy of HER3-DXd in HER3-positive/HR-positive breast cancer by increasing DNA damage. Br J Cancer (2026). https://doi.org/10.1038/s41416-026-03413-1
Image Credits:
AI Generated
DOI:
15 April 2026
Tags: ATR inhibition in breast cancer treatmentATR inhibitors and cancer cell cycle arrestATR kinase role in DNA repaircombination therapy in HR+ breast cancerDNA damage response targeting cancerDNA replication stress in cancer therapyendocrine-resistant hormone receptor-positive breast cancerenhancing HER3-targeted therapy efficacyHER3-DXd antibody-drug conjugatenovel strategies for endocrine resistanceovercoming therapeutic resistance in breast cancertargeted therapy for metastatic breast cancer
{kind=link}