A new study led by Rice University’s Peter Wolynes offers new insights into the evolution of foldable proteins. The research was published in the Proceedings of the National Academy of Sciences.
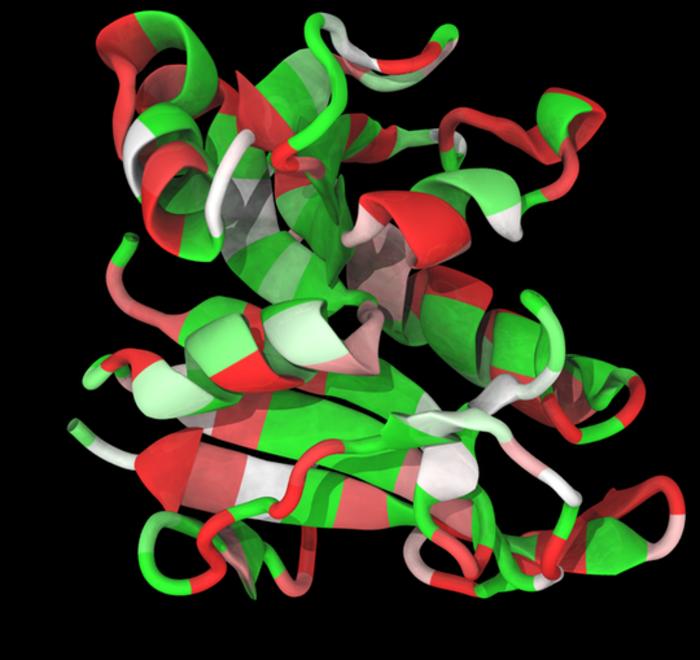
Credit: Image by Carlos Bueno/Rice University.
A new study led by Rice University’s Peter Wolynes offers new insights into the evolution of foldable proteins. The research was published in the Proceedings of the National Academy of Sciences.
Researchers at Rice and the University of Buenos Aires used energy landscape theory to distinguish between foldable and nonfoldable parts of protein sequences. Their study illuminates the ongoing debate about whether the pieces of DNA that code for only part of a protein during their origins can fold on their own.
The researchers focused on the extensive relationship between exons in protein structures and the evolution of protein foldability. They highlighted the significance of exons, the parts of the gene that code for proteins, and introns, the silent regions discarded during gene translation into proteins.
“Using the extensive genomic exon-intron organization and protein sequence data now available, we explored exon boundary conservation and assessed its behavior using energy landscape theoretic measurements,” said Wolynes, the D.R. Bullard-Welch Foundation Professor of Science, professor of chemistry, biosciences, physics and astronomy and co-director of the Center for Theoretical Biological Physics (CTBP).
When genes in pieces were discovered in the 1970s, it was immediately proposed that by breaking up the sequence, this structure helped build foldable proteins. When researchers looked at this again in the 1990s, the existing data was equivocal, Wolynes said.
The team has now assessed exons as potential protein folding modules across 38 abundant and conserved protein families. Over generations, exons can shuffle randomly along the genome, leading to significant changes in genes and the creation of new proteins. The findings indicated deviations in the exon size distribution from exponential decay, suggesting there was evolutionary selection.
“Protein folding and evolution are closely linked phenomena,” said Ezequiel Galpern, a postdoctoral researcher at the University of Buenos Aires.
Natural proteins are linear chains of amino acids that typically fold into compact three-dimensional structures to perform biological functions. The specific sequence of amino acids dictates the final 3D structure. Therefore, the idea that exons translate into independently folded protein regions, or foldons, is very attractive.
Using computational methods, the researchers measured the likelihood of the amino acid chain coded by an exon to fold into a stable 3D structure, similar to the full protein. Their results showed that while not all exons led to foldable modules, the most conserved exons, consistently found in diverse organisms, corresponded with better foldons.
The study found a correlation between protein folding and evolution in certain globular protein families. Protein folding involves amino acid chains folding in space to perform biological functions within relevant timescales. This correlation is a fundamental concept in protein science, assessed using genomic data and energy functions.
Interestingly, the general trend did not hold for all protein families, suggesting that other biological factors may influence protein folding and evolution. The researchers’ work paves the way for future studies to understand these additional factors and their impact on evolutionary biology.
The research team includes Carlos Bueno, a postdoctoral researcher at CTPB; Hana Jaafari, an applied physics graduate student at Rice; and Diego U. Ferreiro, a professor at the University of Buenos Aires.
This study was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas; the Bullard-Welch Chair at Rice; the University of Buenos Aires; NASA Astrobiology Institute; and CTPB.
Journal
Proceedings of the National Academy of Sciences
DOI
10.1073/pnas.2400151121
Article Title
Reassessing the exon–foldon correspondence using frustration analysis
Article Publication Date
2-Jul-2024
{kind=link}