A special issue of the Journal of Huntington’s Disease focusing on recent genetic advances in the disease synthesizes the latest research and has the potential to serve as a catalyst to improve patient care and outcomes
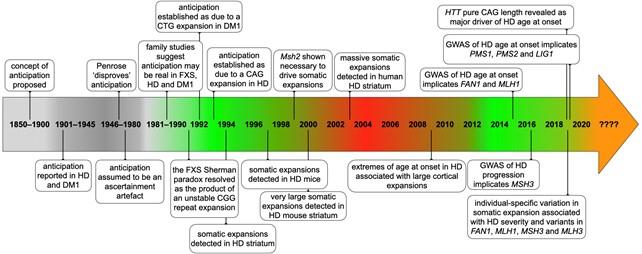
Credit: : Darren G. Monckton in “The Contribution of Somatic Expansion of the CAG Repeat to Symptomatic Development in Huntington’s Disease: A Historical Perspective, ” Journal of Huntington’s Disease 10:1 (February 2021)….
Amsterdam, February 11, 2021 – Recent genetic data from patients with Huntington’s disease (HD) show that DNA repair is an important factor that determines how early or late the disease occurs in individuals who carry the expanded CAG repeat in the HTT gene that causes HD. The processes of DNA repair further expand the CAG repeats in HTT in the brain implicated in pathogenesis and disease progression. This special issue of the Journal of Huntington’s Disease (JHD) is a compendium of new reviews on topics ranging from the discovery of somatic CAG repeat expansion in HD, to our current understanding of the molecular mechanisms involved and the development of potential new therapies targeting these mechanisms.
The CAG expansion varies from 6 to 35 repeats in HTT on chromosomes of unaffected individuals and from more than 36 to greater than 180 repeats in HD patients.
“The CAG repeat mutation in the HTT gene that causes HD was discovered in 1993. Although our understanding of the underlying biology and our ability to model many aspects of the disease have improved substantially, no treatment that alters the course of this devastating disorder has been found,” explain Guest Editors Lesley Jones, PhD (Cardiff University, UK), Vanessa Wheeler, PhD (Massachusetts General Hospital, USA), and Christopher E. Pearson, PhD (The Hospital for Sick Children, University of Toronto, Canada).
Recent advances in genetics have now transformed our understanding of the factors that are critical in the pathogenesis of HD and are beginning to provide similar insight into other repeat expansion disorders such as Friedreich’s ataxia, myotonic dystrophy and many of the repeat-expansion spinocerebellar ataxias. This special issue of JHD provides an important and novel synthesis of the wealth of HD research information and the knowledge of DNA repair and somatic repeat expansion in HD. This has the potential to help drive discovery forward, leading to new treatments and improved quality of life and outcomes for individuals with HD.
The genetic findings presented in this issue examine data in HD, HD models and other diseases and make sense of many previous findings with respect to how DNA repair genes altered the disease in mice. This has highlighted HD biology that can be used to generate new targets for development of drugs to treat the disease. It may eventually allow a better prediction of the clinical course of HD in specific individuals and improve the power of clinical trials enabling shorter trials or trials with fewer participants.
The findings presented in this issue imply that the expanded CAG repeat in HD causes disease through a two-part process. A review entitled “Huntington’s Disease Pathogenesis: Two Sequential Components” outlines how the compelling genetic data that highlight DNA repair and somatic expansion are critically important in the manifestation of HD. It also explores the implications of these data for mechanisms underlying disease pathophysiology. The authors propose that two steps are required for pathogenesis in HD: first an expansion of the HTT CAG repeat in somatic cells, followed by downstream pathogenic events occurring in response to those CAG repeats in cells that are expanded from the inherited length. The authors share access to their website allowing other investigators to explore recent genetic data interactively to support their own research
“The identification of the HD gene has not yet led to an effective treatment, but continued human genetic analysis of genotype-phenotype relationships in large HD subject populations, first at the HTT locus and subsequently genome-wide, has provided insights into pathogenesis that divide the course of the disease into two sequential, mechanistically distinct components,” comments author James F. Gusella, PhD, Massachusetts General Hospital and Harvard Medical School.
Although linking the genetic findings in HD to functional biological systems and then to their actual effect on disease onset or course has been challenging, many pharmaceutical companies, large and small, are now working on the new targets identified by these findings. “Drugging DNA Damage Repair Pathways for Trinucleotide Repeat Expansion Diseases” highlights and addresses the multiple challenges that will need to be overcome to generate new therapeutics or repurposing existing therapeutics that can tackle DNA repair and somatic expansion of the CAG repeat. It illustrates the gaps in our knowledge that we really need to fill to use these findings to generate novel and effective drugs for somatic expansion in HD and potentially other repeat disorders.
“The development of novel DNA damage response (DDR) drugs for neurodegeneration is also facilitated by understanding of challenges with DDR drugs for oncology and key liabilities associated with specific targets,” notes lead author Caroline L. Benn, PhD, LoQus23 Therapeutics. “We need to maintain a watching brief, continue to address the gaps in our understanding, and ensure we continue to work to realize the potential to increase therapeutic benefit and reduce risk. Taken together, we believe success is possible with close collaboration between patients, academic investigators, preclinical drug discoverers, clinicians, diagnostic developers and regulatory bodies.”
“We hope that our colleagues in the global HD research community find this special issue educational and stimulating and that it serves to speed discovery and treatments. We learned a lot – we hope our readers do likewise!” add the Guest Editors.
###
Co-Editors-in-Chief Blair R. Leavitt, MD (The University of British Columbia) and Leslie M. Thompson, PhD (University of California Irvine) have dedicated this special issue in memory of Professor Sir Peter Harper (1939-2021). A visionary geneticist, he was instrumental in the discovery of the repeat mutations causing HD and myotonic dystrophy. “Prof. Harper continued to influence the HD field though his perceptive anticipation of what these genetic discoveries would mean for patients, their families and the direction of HD research. He was a generous and engaged mentor to a whole generation of clinical and non-clinical researchers in HD and other repeat disorders, and his legacy will continue to inspire future generations,” adds Prof. Jones, a long-time colleague and protégé.
Media Contact
Diana Murray
[email protected]
Related Journal Article
http://dx.

{kind=link}