Study finds a short section of the amyloid beta protein is recognized by the cellular prion protein, which mediates its uptake into neurons and subsequent toxicity
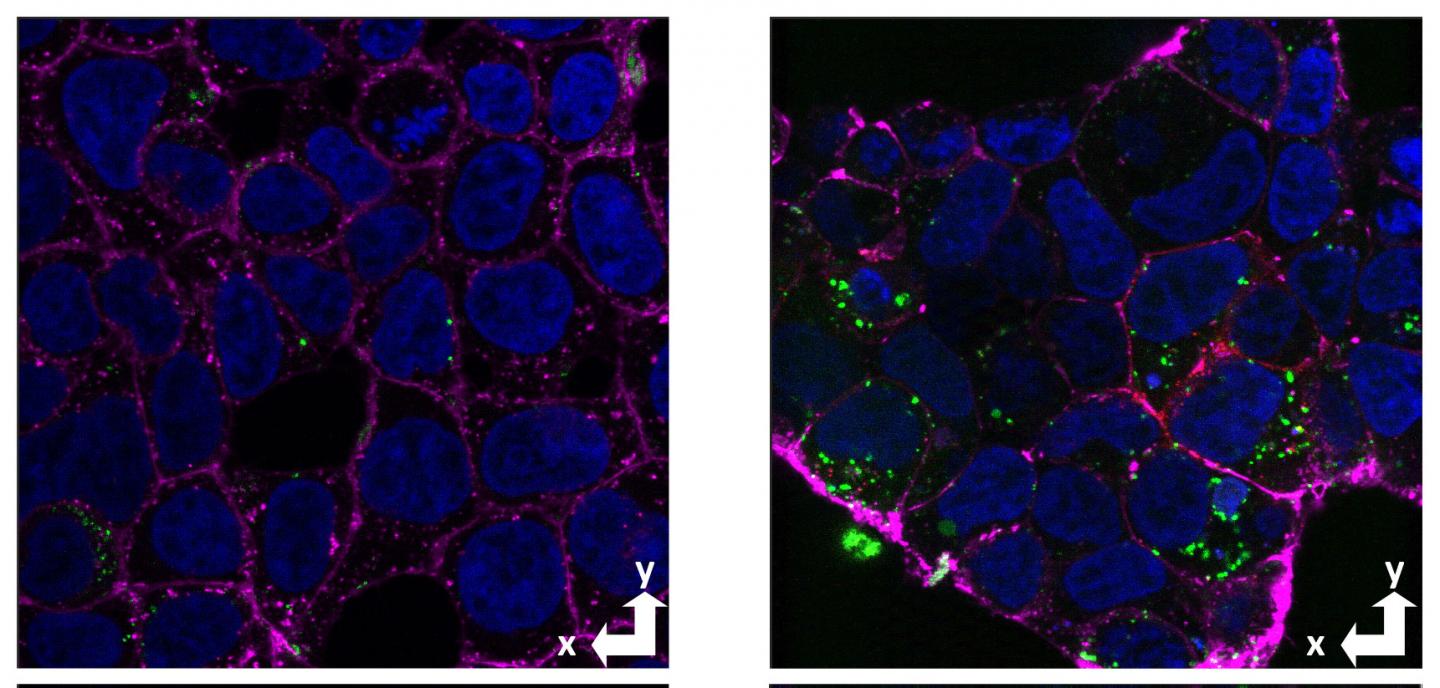
Credit: Graham Roseman, UCSC
One of the hallmarks of Alzheimer’s disease is the formation of amyloid plaques–sticky clumps of a protein called amyloid beta–that collect between neurons in the brain. Increasingly, however, attention has turned from these insoluble plaques to soluble forms of amyloid beta that can be taken up into neurons and are highly neurotoxic.
A new study by researchers at the University of California, Santa Cruz, pinpoints a segment of the amyloid beta protein that is recognized by receptors involved in neuronal uptake of this toxic peptide. The researchers used a novel approach to study the mechanisms of cellular uptake of amyloid beta. Their findings, published November 2 in Proceedings of the National Academy of Sciences, suggest that targeting this process may be a promising approach for Alzheimer’s drug development.
“There are many different ways that amyloid beta can be toxic inside cells, so wouldn’t it be nice if we could block its uptake by neurons? This is a pathway we can target,” said corresponding author Jevgenij Raskatov, assistant professor of chemistry and biochemistry at UC Santa Cruz.
In the new study, Raskatov teamed up with co-corresponding author Glenn Millhauser, distinguished professor of chemistry and biochemistry at UCSC, to study the interactions of amyloid beta with the cellular prion protein. Millhauser’s lab studies the structure and function of the prion protein, a membrane protein found on the surfaces of various cell types, including neurons in the brain.
Previous studies by other researchers have indicated that the normal cellular prion protein (not the abnormal variant that causes prion diseases) binds to clumps of amyloid beta and is involved in uptake and neurotoxicity. The UCSC researchers, led by graduate student Alejandro Foley and postdoctoral researcher Graham Roseman, sought to test whether the prion protein also acts as a receptor to take up soluble forms of amyloid beta, and to identify the site within amyloid beta that binds to this receptor.
The researchers took an approach based on previous work from Raskatov’s lab using mirror-image versions of amyloid beta to show that cellular uptake is mostly mediated by receptors on the cell surface. In the earlier work, researchers compared the uptake of natural amyloid beta with a synthetic version in which the arrangement of the atoms in the protein’s amino acids is a mirror image of the natural arrangement. This rearrangement subtly changes the protein’s structure in ways that would interfere with its binding to a receptor, so the finding that cellular uptake of the mirror-image version was greatly reduced pointed to receptor-mediated uptake.
Amyloid beta can be different lengths, but the most toxic variant is 42 amino acids long. To isolate the site involved in receptor binding, the researchers created a library of peptide segments of amyloid beta composed of, for example, just amino acids 1 through 16 or 1 through 30. For each segment, they made a version with natural amino acids (the “L stereoisomer”) and one with mirror-image amino acids (the “D stereoisomer”).
After testing all the peptides in the library for cellular uptake, they found that amino acids 1-30 showed the same stereoselectivity as full-length amyloid beta, with much greater uptake of the L form than the D form. In addition, this segment is completely soluble and does not form aggregates because it is missing a long hydrophobic domain involved in the aggregation of amyloid beta into clumps and fibrils.
“With this shortened amyloid beta, we are able to decouple cellular uptake from aggregation, giving us a great model for studying uptake,” Raskatov said.
By assessing amyloid beta uptake in cells with and without the prion protein, the researchers demonstrated for the first time the prion protein’s role in cellular uptake of soluble amyloid beta, consistent with its selectivity for the L stereoisomer of amyloid beta.
The non-aggregating amyloid beta segment 1-30 showed the same prion-dependent uptake and stereoselectivity. The researchers also used nuclear magnetic resonance spectroscopy to gather detailed information about the interactions involved in the binding of amyloid beta to the cellular prion protein.
Their findings show that the binding of amyloid beta at the cell surface, leading to its internalization, is largely due to the amino acid sequence 1-30 and not the state of aggregation. When amyloid beta molecules begin to aggregate, they form “oligomers” consisting of a small number of molecules stuck together that are still soluble and can be taken up by neurons. These soluble oligomers are increasingly considered to be the form of amyloid beta that triggers the pathological processes leading to Alzheimer’s disease, but there are many different aggregated forms.
“There are different levels of aggregation, different sizes and types of oligomers, and a big question has been which forms are internalized and cause neurotoxicity,” Raskatov said. “Our findings suggest that any form in which that sequence of amino acids is exposed will bind to the prion protein.”
According to Millhauser, “The initial steps leading to Alzheimer’s disease may be the prion protein-mediated transport of soluble amyloid beta into neurons, where it then clumps, forming toxic aggregates that ultimately lead to the characteristic plaques associated with the disease.”
Although the prion protein seems to be responsible for most of the amyloid beta uptake, the study suggests other receptors may offer alternate routes. Nevertheless, drugs that target the 1-30 segment of amyloid beta or the sites it binds to on receptors may hold therapeutic potential for treating Alzheimer’s disease.
“Our findings open up new avenues for understanding Alzheimer’s and suggest promising strategies for therapeutics,” Millhauser said.
###
In addition to Raskatov, Millhauser, Foley, and Roseman, the coauthors include Ka Chan, Amanda Smart, Thomas Finn, Kevin Yang, and Scott Lokey, all at UC Santa Cruz. This work was funded by the National Institutes of Health.
Media Contact
Tim Stephens
[email protected]
Related Journal Article
http://dx.

{kind=link}