Results show that retinitis pigmentosa 59 may not be a congenital disorder of glycosylation, as was long assumed

Credit: UAB
BIRMINGHAM, Ala. – Researchers who made a knock-in mouse-model of the genetic disorder retinitis pigmentosa 59, or RP59, expected to see retinal degeneration and retinal thinning. As reported in the journal Cells, they surprisingly found none, calling into question the commonly accepted — though never proved — mechanism for RP59.
“Our findings bring into question the current concept that RP59 is a member of a large and diverse class of diseases known as ‘congenital disorders of glycosylation,'” said Steven Pittler, Ph.D., professor and director of the University of Alabama at Birmingham School of Optometry and Vision Science Vision Science Research Center. “While in principle it would be reasonable to consider RP59 as a congenital disorder of glycosylation, due to the associated mutation in DHDDS, an enzyme required for glycosylation, there is no direct evidence to demonstrate a glycosylation defect in the human retinal disease or in any animal model of RP59 generated to date.”
This means the mechanism of DHDDS-dependent retinal degeneration in human RP59 patients remains unknown, and appears to be more complex than just a DHDDS loss of function.
Retinitis pigmentosa is a group of rare genetic disorders affecting the light-sensitive retinal tissue at the back of the eye. Patients first notice night blindness and loss of peripheral vision, and the disease can progress to vision loss and legal blindness. RP59 has changes in the gene for DHDDS, one of the 50 genes that can lead to RP. RP59 is found in one in 100 Ashkenazi Jewish people and one in 2,009 people worldwide.
In RP59, the lysine that is the 42nd amino acid of the DHDDS enzyme is changed to a glutamic acid, a change known as a K42E point mutation. So Pittler and colleagues at UAB and the State University of New York at Buffalo, or SUNY-Buffalo, examined the retinas of mice that were made homozygous for a DHDDS K42E mutation.
Using spectral domain-optical coherence tomography, they found no evidence of retinal degeneration, even up to one year of age. Also, there was no evidence of compromised protein N-glycosylation, meaning there was no significant DHDDS loss of function in the K42E mice. Opsin, a protein in photoreceptor cells involved in vision, is often mislocalized when photoreceptor cells degenerate; however, the K42E mice showed no change in opsin localization.
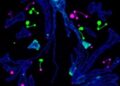
What the researchers did find was extensive gliosis in the retinas of the mutant mice, which was spread in a radial pattern throughout the inner retinal layers to the outer plexiform layer of the retina. There was also intense gliosis at the vitreoretinal interface. Gliosis is a change in glial cells of the central nervous system, often in response to injury. In the retina, it affects glial M?ller cells, and that reactive gliosis can have harmful effects on vision.
“These results indicate massive gliotic activation,” Pittler said, “which is remarkable considering the lack of overt retinal degeneration or loss of retinal neurons.”
###
The study, “Lack of overt retinal degeneration in a K42E Dhdds knock-in mouse model of RP59,” is published in Cells. Co-authors with Pittler are Sriganesh Ramachandra Rao and Steven J. Fliesler, SUNY-Buffalo; and Pravallika Kotla and Mai N. Nguyen, UAB Department of Optometry and Vision Science.
Support came from National Institutes of Health grants EY029341 and P30 003039; a Fight for Sight Summer Student Fellowship, a Career-Starter Research Grant from the Knights Templar Eye Foundation, the UAB Vision Science Research Center and the Veterans Affairs Western New York Healthcare System.
Pittler and colleagues have also published two companion papers that further probe the role of DHDDS in RP.
The first study, “Selective ablation of dehydrodolichol diphosphate synthase in murine retinal pigment epithelium causes RPE atrophy and retinal degeneration,” is published in Cells. It showed that retinal pigment epithelium-specific deletion of the gene for DHDDS in mice induced structural and functional deficits in the retinal pigment epithelium and the photoreceptors. This suggests that retinal pigment epithelium pathology may be a significant contributor to the retinal degeneration observed in humans with RP59 mutations.
Researchers at UAB and SUNY-Buffalo did the study.
The second study, “Retinal degeneration caused by rod-specific Dhdds ablation occurs without concomitant inhibition of protein N-glycosylation,” is published in IScience. It examined the rod photoreceptor-specific ablation of the gene for DHDDS. The researchers found that mouse retinas had mild retinal dysfunction at four weeks after birth, followed by rapid photoreceptor degeneration and almost complete loss of rods and cones by six weeks after birth. Rods and cones are the photoreceptors of the retina. Retinal proteins, including opsin, still showed N-glycosylation, which again challenges the conventional mechanistic view of RP59 as a congenital disorder of glycosylation.
Researchers at UAB, SUNY-Buffalo and the Polish Academy of Sciences did the study.
Media Contact
Jeff Hansen
[email protected]
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}