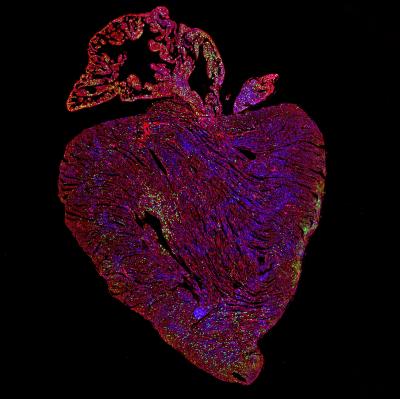
Credit: Dr Cathy Wilson, University of Cambridge
Researchers trying to turn off a gene that allows cancers to spread have made a surprising U-turn. By making the gene overactive and functional in the hearts of mice, they have triggered heart cell regeneration. Since adult hearts cannot usually repair themselves once damaged, harnessing the power of this gene represents major progress towards the first curative treatment for heart disease.
“This is really exciting because scientists have been trying to make heart cells proliferate for a long time. None of the current heart disease treatments are able to reverse degeneration of the heart tissue – they only slow progression of the disease. Now we’ve found a way to do it in a mouse model,” said Dr Catherine Wilson, a researcher in the University of Cambridge’s Department of Pharmacology, who led the study.
The cell cycle – through which cells make copies of themselves – is tightly controlled in mammalian cells. Cancer develops when cells start to replicate themselves uncontrollably, and the Myc gene plays a key role in the process. Myc is known to be overactive in the vast majority of cancers, so targeting this gene is one of the highest priorities in cancer research. Much recent research has focused on trying to take control of Myc as a means of cancer therapy.
When the researchers made Myc overactive in a mouse model, they saw its cancerous effects in organs including the liver and lungs: huge amount of cells started replicating over the course of a few days. But in the heart, nothing much happened.
They found that Myc-driven activity in heart muscle cells is critically dependent on the level of another protein called Cyclin T1, made by a gene called Ccnt1, within the cells. When the Ccnt1 and Myc genes are expressed together, the heart switches into a regenerative state and its cells start to replicate. The results are published today in the journal Nature Communications.
“When these two genes were overexpressed together in the heart muscle cells of adult mice we saw extensive cell replication, leading to a large increase in the number of heart muscle cells,” said Wilson.
Heart failure affects around 23 million people worldwide each year, and there is currently no cure. After a heart attack, an adult human heart can lose up to one billion heart muscle cells – called cardiomyocytes. Unlike many other organs in the body, the adult heart can’t regenerate itself, so these cells are never replaced. Their loss reduces the strength of the heart and causes scar formation, heart failure and ultimately death.
Using a next generation sequencing technology called ChIP, the researchers were able to watch the action of Myc in the heart cells. Myc produces a protein – called a transcription factor – that binds to the DNA in specific cells and activates gene expression. But despite the protein binding successfully, the heart cells didn’t start to replicate themselves because the protein could not activate gene expression. Another protein vital to gene expression, Cyclin T1, was deficient in the heart. Adding it to the cells with the overactive Myc caused the cells to start proliferating.
“None of the current treatment options can reverse the degeneration of heart tissue. The inability of the heart to regenerate itself is a significant unmet clinical need,” said Wilson. “We found that even when Myc is switched on in a heart, the other tools aren’t there to make it work, which may be one of the reasons heart cancer is so extremely rare. Now we know what’s missing, we can add it and make the cells replicate.”
As the world’s population grows and the prevalence of heart failure increases, the cost of patient care is anticipated to increase dramatically. The researchers hope to develop their finding into a genetic therapy for the treatment of heart disease. “We want to use short-term, switchable technologies to turn on Myc and Cyclin T1 in the heart. That way we won’t leave any genetic footprint that might inadvertently lead to cancer formation,” said Wilson.
###
Media Contact
Jacqueline Garget
[email protected]
Related Journal Article
http://dx.



{kind=link}