A groundbreaking study recently published in Genes & Diseases has shed new light on the clinical complexities linked to loss-of-function mutations in the IKBKG gene, also known as NEMO. This gene encodes a crucial regulatory protein within the NF-κB signaling pathway, a key cellular mechanism responsible for orchestrating immune responses, inflammation, and cell survival. Intriguingly, mutations impairing this gene’s function manifest in a spectrum of rare, often devastating disorders such as Incontinentia Pigmenti (IP), Anhidrotic Ectodermal Dysplasia with Immunodeficiency (EDA-ID), isolated Immunodeficiency (ID), and NEMO Deleted Exon 5 Autoinflammatory Syndrome (NDAS). This extensive review offers an unprecedented deep dive into the genotype-phenotype correlations that have long confounded clinicians and researchers alike.
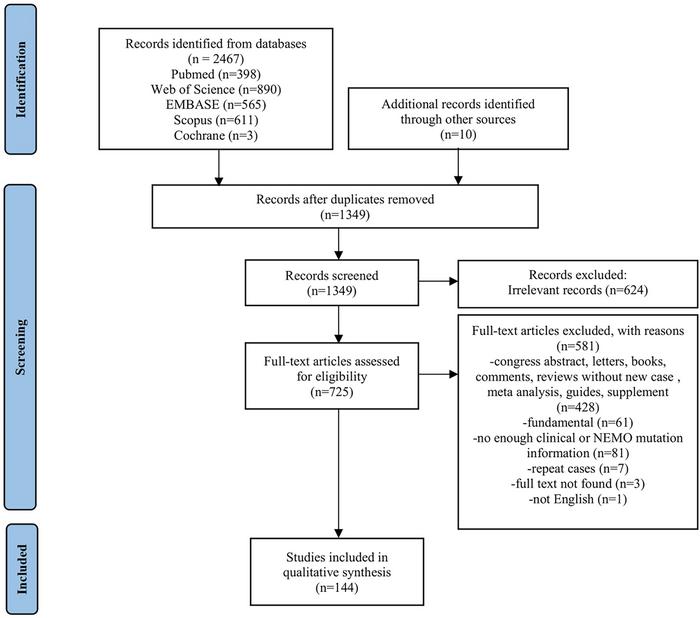
The study’s authors undertook a rigorous analysis of data compiled from 144 publications, collectively documenting clinical presentations of 564 patients harboring IKBKG mutations. Their findings reveal a striking sex-based disparity in phenotype expression: approximately 78% of cases aligned with Incontinentia Pigmenti, a condition predominantly affecting females due to its X-linked dominant inheritance pattern and the male lethality often associated with full loss-of-function mutations. Conversely, EDA-ID, ID, and NDAS overwhelmingly presented in male patients, accounting for 100% male prevalence in these groups. This dichotomy underscores the pivotal role of X chromosome biology in disease manifestation, further complicating clinical diagnosis and patient management.
Incontinentia Pigmenti, the most prevalent clinical presentation, manifests with hallmark dermatological abnormalities, observed in nearly 90% of cases. These cutaneous signs vary from blistering and hyperpigmentation in infancy to atrophic scarring in later life stages. The study elucidates that female carriers typically survive due to cellular mosaicism afforded by X-chromosome inactivation, which mitigates complete gene loss. However, the molecular etiology remains intricate, with phenotypic expression influenced by the specific mutation type and affected protein domains, reflecting a delicate balance within the NF-κB pathway’s regulatory network.
.adsslot_sLjaJzxlIk{ width:728px !important; height:90px !important; }
@media (max-width:1199px) { .adsslot_sLjaJzxlIk{ width:468px !important; height:60px !important; } }
@media (max-width:767px) { .adsslot_sLjaJzxlIk{ width:320px !important; height:50px !important; } }
ADVERTISEMENT
The EDA-ID phenotype, closely linked to immune dysfunctions, was marked by a high incidence of dental anomalies—approximately 68.5% of cases—indicative of developmental disruptions in ectodermal derivatives. Beyond structural abnormalities, the immunodeficiency associated with EDA-ID manifests profoundly as patients exhibit recurrent infections, primarily driven by bacterial pathogens such as Mycobacterium and Streptococcus species. Importantly, the study highlights that all male patients presenting with EDA-ID or isolated Immunodeficiency suffered from debilitating infections, emphasizing the gene’s vital immunological role.
At the molecular level, certain mutations exhibit dual phenotypic associations. For example, the frameshift mutation E390RfsX5 has been identified to cause both IP and EDA-ID, suggesting that even subtle alterations in IKBKG function can precipitate vastly different clinical courses. Likewise, the H413R missense mutation correlates with extensive immune system compromise. These findings reflect the nuanced genotype-phenotype relationships mediated by the specific structural domains within the NEMO protein, particularly the zinc finger domain, which bears critical importance for protein-protein interactions essential to NF-κB signaling.
Notably, a substantial subset of patients with EDA-ID demonstrated hypogammaglobulinemia, with reduced immunoglobulin G (IgG) levels and occasional hyper-IgM syndrome, a paradoxical immunological profile wherein class-switch recombination is impaired. This immunodeficiency results in a compromised humoral immunity, rendering patients susceptible to opportunistic infections and underscoring the need for vigilant clinical monitoring and early therapeutic interventions such as immunoglobulin replacement therapy.
The study also brings to the forefront the zinc finger (ZF) domain of the NEMO protein as a hotspot for mutations predisposing to severe, life-threatening phenotypes. This structural motif is instrumental in ubiquitin-binding and the assembly of signaling complexes necessary for NF-κB activation. Disruption of the ZF domain effectively abrogates signal transduction, leading to profound immunodeficiency and aberrant inflammatory responses. These mechanistic insights underscore the domain’s essential role in maintaining immune homeostasis.
One of the most challenging aspects illuminated by this research is the considerable heterogeneity observed within clinical presentations, even among individuals carrying identical mutations. This phenotypic variability suggests that other genetic modifiers, environmental factors, or epigenetic mechanisms might modulate disease severity and progression. Consequently, a one-size-fits-all diagnostic or therapeutic approach proves inadequate, necessitating personalized medicine strategies that consider the unique genetic and clinical context of each patient.
The early detection of IKBKG mutations emerges as a critical factor in optimizing patient outcomes. Although clinical symptoms frequently manifest in infancy or early childhood, the study highlights delays in genetic diagnosis as a significant barrier to timely management. Clinical vigilance combined with comprehensive genetic screening, especially in patients exhibiting recurrent infections, unexplained inflammatory conditions, or ectodermal dysplasia, could enable prompt intervention, mitigate complications, and improve quality of life.
Intriguingly, the study advocates for revising the diagnostic criteria for Incontinentia Pigmenti to incorporate central nervous system (CNS) abnormalities, which have been underrecognized in previous clinical frameworks. Neurological manifestations such as seizures, developmental delay, and cerebral infarcts are more prevalent than previously appreciated and may hold prognostic significance. Integrating CNS features into diagnostic protocols could enhance early recognition and facilitate multidisciplinary care approaches.
Given the complexity and phenotypic spectrum of IKBKG-related disorders, the authors underscore the urgent need for next-generation targeted therapies aimed at modulating the NF-κB pathway. Such precision medicine approaches might involve gene therapy, small molecule inhibitors, or biologics designed to restore or compensate for aberrant signaling. The development of these novel interventions hinges on an intimate understanding of the molecular underpinnings detailed in this extensive review.
This seminal research marks a pivotal advancement in the field of immunogenetics by compiling a vast dataset that bridges molecular genetics with clinical medicine. It provides an invaluable resource for clinicians, genetic counselors, and researchers striving to unravel the complexities of NEMO/IKBKG-associated diseases. Through elucidating the intricate genotype-phenotype relationships and calling attention to early diagnostic and therapeutic strategies, this work paves the way toward better management and improved prognosis for affected individuals worldwide.
In conclusion, with an impact factor of 9.4 and a Scopus CiteScore of 8.4, Genes & Diseases continues to publish transformative studies like this that deepen our comprehension of molecular pathogenesis. The widespread implications of this research extend beyond rare genetic disorders, as NF-κB pathway dysfunctions are implicated broadly across immunology, oncology, and inflammatory diseases. The scientific community eagerly anticipates follow-up investigations that explore therapeutic possibilities and refine disease classification, harnessing the potential to revolutionize patient care in this challenging domain.
Subject of Research: Clinical implications of loss-of-function mutations in IKBKG/NEMO gene affecting the NF-κB signaling pathway.
Article Title: Clinical relevance of loss-of-function mutations of NEMO/IKBKG
News Publication Date: 2025
References:
Jin Wang, Kexin Shen, Hongxia Lou, Lina Zhou, Yunfei An, Xiaodong Zhao, Yuan Ding, Clinical relevance of loss-of-function mutations of NEMO/IKBKG, Genes & Diseases, 2025, 101531, DOI: 10.1016/j.gendis.2025.101531
Image Credits: Genes & Diseases
Keywords: Cancer genetics
Tags: Anhidrotic Ectodermal Dysplasiaautoinflammatory syndromesclinical impact of gene mutationsgenotype-phenotype correlationsIKBKG mutationsIncontinentia PigmentiNEMO geneNF-κB signaling pathwaypatient data analysis in geneticsrare immunodeficiency syndromessex-based disparity in immunodeficiencyX-linked dominant inheritance


{kind=link}