Credit: Wei Hao/Princeton
The same algorithms that personalize movie recommendations and extract topics from oceans of text could bring doctors closer to diagnosing, treating and preventing disease on the basis of an individual's unique genetic profile.
In a study to be published Monday, Nov. 7 in Nature Genetics, researchers at Columbia and Princeton universities describe a new machine-learning algorithm for scanning massive genetic data sets to infer an individual's ancestral makeup, which is key to identifying disease-carrying genetic mutations.
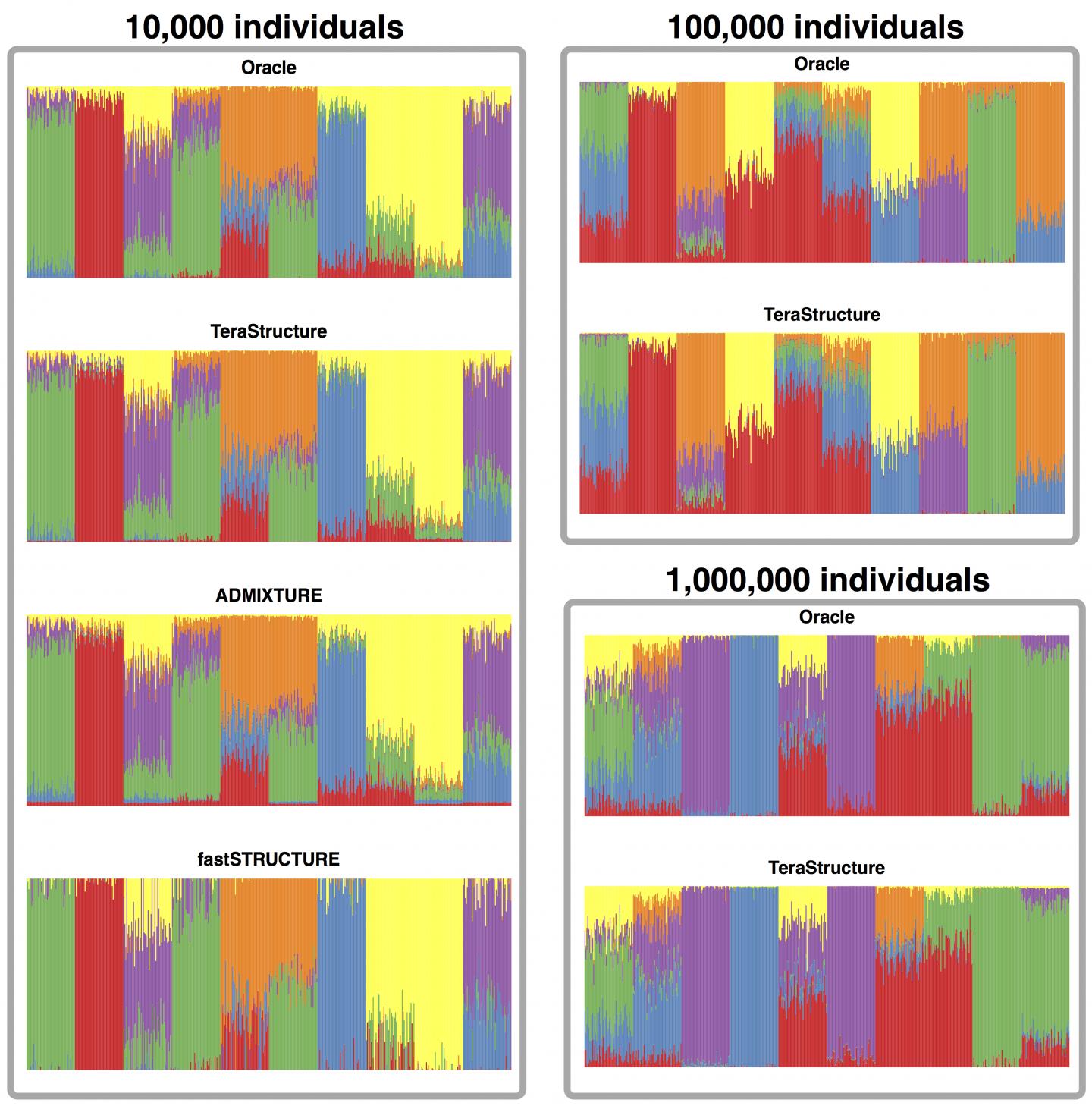
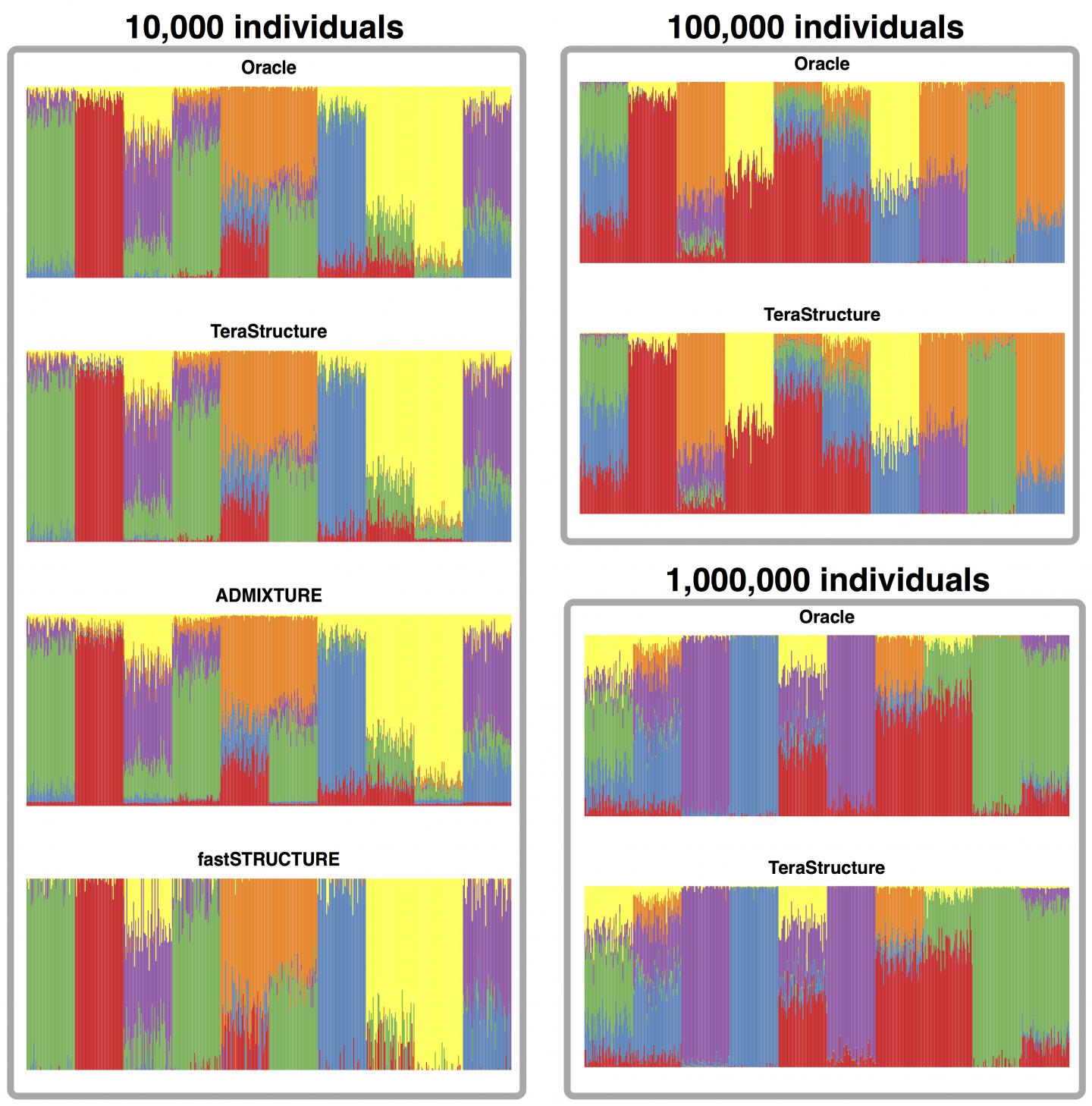
On simulated data sets of 10,000 individuals, TeraStructure could estimate population structure more accurately and twice as fast as current state-of-the art algorithms, the study said. TeraStructure alone was capable of analyzing 1 million individuals, orders of magnitude beyond modern software capabilities, researchers said. The algorithm could potentially characterize the structure of world-scale human populations.
"We're excited to scale some of our recent machine learning tools to real-world problems in genetics," said David Blei, a professor of computer science and statistics at Columbia University and member of the Data Science Institute.
The cost of genetic sequencing has fallen sharply since the first complete mapping of the human genome in 2003. More than a million people now have sequenced genomes, and by 2025 that number could rise to 2 billion.
The technology to put this data into context, however, has lagged and remains one of the barriers to tailoring healthcare to an individual's DNA. To identify disease-causing variants in a genome, one of the goals of personalized medicine, researchers need to know something about his or her ancestry to control for normal genetic variation within a subpopulation.
"We can run software on a few thousand people, but if we increase our sample size to a few hundred thousand, it can take months to infer population structure," said Kai Wang, director of clinical informatics at Columbia's Institute for Genomic Medicine, who was not involved in the study. "This new tool addresses these limitations, and will be very useful for analyzing the genomes of large populations."
The researchers' algorithm, called TeraStructure, builds on the widely used and adapted STRUCTURE algorithm first described in the journal Genetics in 2000. The STRUCTURE algorithm cycles through an entire data set, genome by genome, one million variants at a time, before updating its model to both characterize ancestral populations and estimate their proportion in each individual. The model gets refined after repeated passes through the data set.
TeraStructure, by contrast, updates the model as it goes. It samples one genetic variant at one location, and compares it to all variants in the data set at the same location across the data set, producing a working estimate of population structure. "You don't have to painstakingly go through all the points each time to update your model," said Blei.
STRUCTURE is mathematically similar to a topic-modeling algorithm Blei developed independently in 2003 that made it possible to scan large numbers of documents for overarching themes. Blei's algorithm and its underlying LDA model have been used, among other things, to analyze published research in the journal Science to understand the evolution of scientific ideas and review regulatory meeting transcripts for insight into how the U.S. Federal Reserve sets interest rates.
More recently, Blei has experimented with statistical techniques to extend probabilistic models to massive data sets. One technique, stochastic optimization, developed in 1951 by statistician Herbert Robbins just before arriving at Columbia, uses a small, random subset of observations to compute a rough update for the model's parameters.
Continuously refining the model with each new observation, stochastic optimization algorithms have been enormously successful in scaling up machine learning approaches used in deep learning, recommendation systems and social network analysis.
In a 2010 paper, Online Learning for LDA, Blei and his colleagues applied stochastic optimization to Blei's earlier LDA model. In a later paper, Stochastic Variational Inference, they showed that stochastic optimization could be applied to a range of models. As Matthew Hoffman, a coauthor of both papers, now a senior research scientist at Adobe Research explains, "Stochastic optimization algorithms often find a good solutions before they've even analyzed the whole dataset."
In the Nature Genetics study, they apply these ideas to the STRUCTURE method. In their analysis of two real-world data sets–940 individual genomes from Stanford's Human Genome Diversity Project and 1,718 genomes from the 1000 Genomes Project–they found that TeraStructure performed comparably to the more recent ADMIXTURE and fastSTRUCTURE algorithms.
But when they ran TeraStructure on a simulated data set of 10,000 genomes, it was more accurate and two to three times faster at estimating population structure, the study said. The researchers also showed that TeraStructure alone could analyze data sets as large as 100,000 genomes and 1 million genomes.
Matthew Stephens, a genetics researcher at University of Chicago who helped develop the STRUCTURE algorithm, called TeraStructure's performance impressive. "I think these results will motivate future applications of this kind of algorithm in challenging inferences problems," he said
The study also received praise from other researchers working with big genetic data sets. "We now have the technology to create the data," said Itsik Pe'er, a computational geneticist at Columbia Engineering who was not involved in the study. "But this paper really allows us to use it."
###
The study is titled, "Scaling probabilistic models of genetic variation to millions of humans." Other authors are Prem Gopalan, Wei Hao, and John Storey, of Princeton.
The Data Science Institute at Columbia University is training the next generation of data scientists and developing innovative technology to serve society. http://datascience.columbia.edu/
Media Contact
Kim Martineau
[email protected]
646-717-0134
@columbia
http://www.columbia.edu

{kind=link}