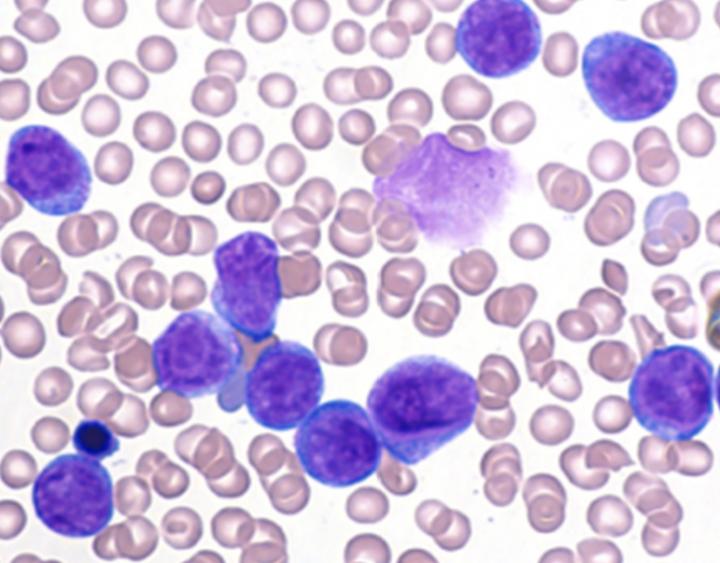
Credit: Behrens et al., 2017
Researchers in Germany have discovered that a tumor suppressor protein thought to prevent acute myeloid leukemia (AML) can actually promote a particularly deadly form of the disease. The study, "RUNX1 cooperates with FLT3-ITD to induce leukemia," which will be published online February 17 in The Journal of Experimental Medicine, suggests that targeting this protein could be an effective treatment for certain AML patients.
AML accounts for over 1 percent of all cancer deaths in the United States and is characterized by an excessive proliferation of hematopoietic stem cells in the bone marrow and their subsequent failure to differentiate into white blood cells. AML can be caused by various combinations of gene mutations. One of the most common mutations is in the gene encoding the cell surface signaling protein FLT3, and patients with this mutation show poor rates of survival. The mutant form of FLT3 can promote cell proliferation, but experiments in mice have shown that it isn't sufficient to block white blood cell differentiation and induce AML on its own.
Carol Stocking and colleagues at the Heinrich-Pette-Institute, Leibniz Institute for Experimental Virology in Hamburg noticed that many patients carrying the mutant form of FLT3 also showed increased levels of a transcription factor called RUNX1. "This was unexpected because up to 20 percent of AML patients carry mutations that inactivate RUNX1, which is generally considered to be a tumor suppressor that prevents the formation of leukemias," Stocking says.
Stocking's team found that reducing RUNX1 levels attenuated the ability of human AML cells expressing mutant FLT3 to form tumors when injected into mice. In contrast, elevated RUNX1 levels worked with mutant FLT3 to induce AML. Mouse hematopoietic stem cells expressing mutant FLT3 were highly proliferative, and co-expression of RUNX1 blocked their differentiation, allowing them to give rise to AML.
Mutant FLT3 appears to stabilize and activate RUNX1 by promoting the transcription factor's phosphorylation. Active RUNX1 then blocks white blood cell differentiation, at least in part, by inducing another transcription factor called Hhex. Hematopoietic stem cells expressing both Hhex and mutant FLT3 also gave rise to AML, the researchers found.
RUNX1 may therefore suppress the initiation of AML but, after being activated by mutant FLT3, block white blood cell differentiation and promote tumor development. "Therapies that can reverse this differentiation block may offer significant therapeutic efficacy in AML patients with FLT3 mutations," says Stocking. "Ablating RUNX1 is toxic to leukemic cells but not to normal hematopoietic stem cells, so inhibiting RUNX1 may be a promising target in combination with FLT3 inhibitors."
###
Behrens et al., 2017. J. Exp. Med. http://jem.rupress.org/cgi/doi/10.1084/jem.20160927?PR
About The Journal of Experimental Medicine
The Journal of Experimental Medicine (JEM) features peer-reviewed research on immunology, cancer biology, stem cell biology, microbial pathogenesis, vascular biology, and neurobiology. All editorial decisions are made by research-active scientists in conjunction with in-house scientific editors. JEM provides free online access to many article types from the date of publication and to all archival content. Established in 1896, JEM is published by The Rockefeller University Press. For more information, visit jem.org.
Visit our Newsroom, and sign up for a weekly preview of articles to be published. Embargoed media alerts are for journalists only.
Follow JEM on Twitter at @JExpMed and @RockUPress.
Media Contact
Ben Short
[email protected]
212-327-7053
@RockUPress
http://www.rupress.org/
############
Story Source: Materials provided by Scienmag

{kind=link}