Stampede2 and Comet systems complete simulations pertinent to coronavirus, DNA replication
Credit: Mandal et al.
Fundamental research supported by supercomputers could help lead to new strategies and better technology that combats infectious and genetic diseases.
Viruses such as the dreaded severe acute respiratory syndrome coronavirus 2 rely on the host cell membrane to drastically bend and eventually let loose the replicated viruses trapped inside the cell. Scientists have used supercomputer simulations to help propose a mechanism for this budding off of viruses. What’s more, a related study also used simulations to find a mechanism for how the DNA of all life adds a base to its growing strand during replication..
The study on cell membrane remodeling, important for viral reproduction, cell growth and communication, and other biological processes was published online in the Biophysical Journal in February 2020. The study co-author Qiang Cui also was part of a study on DNA base addition, published in the Proceedings of the National Academy of Sciences, December 2019. Qiang Cui is a professor in the Departments of Chemistry, Physics, and Biomedical Engineering, Boston University.
Cui is also the principal investigator on both studies for supercomputer time awarded through XSEDE, the Extreme Science and Engineering Discovery Environment funded by the National Science Foundation. “Supercomputers with massive parallelization are very much required to push the boundary of bimolecular simulations,” Cui said.
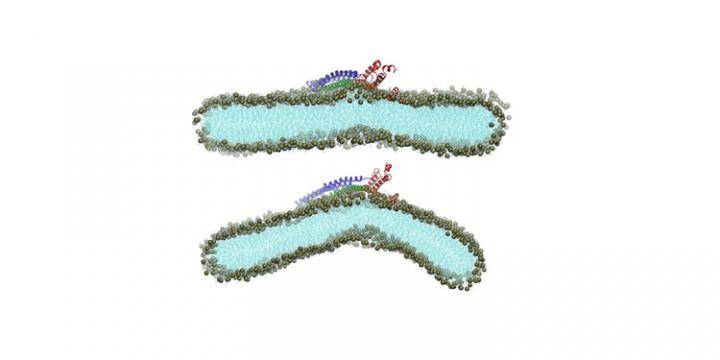
His science team developed supercomputer simulations of the cell membrane, in particular filaments of the Vps32 protein, a major component of the endosomal sorting required for transport complex (ESCRT-III), which was the prime suspect for the driving force that causes the cell membrane to form buds in a process called membrane invagination. ESCRT proteins function in the cytosol, the liquid inside cells surrounding organelles, the cell subunits. They perform various jobs such as making organelles; sorting recyclable material in the cell and ejecting waste, and more.
Electron microscopy shows the Vps32 protein polymerizes, or assembles itself into a corkscrew shape during membrane invagination. The study authors sought to establish whether the atomistic forces inside Vps32 cause it to bend and twist, ultimately tugging and budding off the membrane. Unfortunately, experimental studies currently lack the resolution to characterize the protein-membrane interactions that lead to the membrane deformations.
The science team employed atomistic molecular dynamics simulations to investigate protein-protein interfaces in one-dimensional filament structures in solution and also to find the residues holding the filament together. They also studied the protein-membrane interface using a Vps32 trimer model.
“I think the most interesting observation is that the ESCRTIII polymer that we studied features a clear intrinsic twist,” Cui said. “This suggests that twisting stress that accumulates as the polymer grows on the surface might play a major role in creating the three-dimensional buckling of the membrane. People focused more on the bending of the filament in the past.”
“We also showed explicitly that the N-terminal helix does generate explicit curvature,” Cui added. “People speculated about this before, since amphipathic helices are known to do so in other systems.”
Amphipathic molecules contain both water-loving (hydrophilic) and water-hating (hydrophobic) parts. “Nevertheless, explicitly showing the curvature generated by atomistic forces is important because even more recent studies appear to argue that Vps32 alone is unable to generate membrane curvature,” Cui said. The proposed mechanism supported by simulations basically involves initially dimpling and then pushing out of the membrane as the corkscrew Vps32 protein filament grows, eventually causing the neck of the membrane invagination.
Simulations of systems containing up to two million atoms posed a large hurdle for Cui and his colleagues. They applied for and were awarded supercomputing time through XSEDE, and completed their simulations on the Stampede2 system at the Texas Advanced Computing Center of UT Austin.
“Stampede2 has been crucial for us to set up these relatively large-scale membrane simulations,” Cui said.
While this study is pure research, the knowledge gained could help benefit society. “Membrane remodeling is an important process that underlies many crucial cellular functions and events, such as synaptic transmission and virus infection. Understanding the mechanism of membrane remodeling will help propose new strategies for battling human diseases due to impaired membrane fusion activities — or preventing viral infection — a timely topic these days given the quick spread of the new coronavirus,” Cui said.
Cui also co-authored a computational study that used supercomputer simulations to determine a chemical mechanism for the reaction of nucleotide addition, used in the cell to add nucleotide bases to a growing strand of DNA.
“By doing that, computationally, we are also able to determine the role of a catalytic metal ion of magnesium that’s in the active site of the enzyme DNA polymerase,” said study co-author Daniel Roston, an assistant project scientist in the Department of Chemistry and Biochemistry at UC San Diego. “This metal has been a bit controversial in the literature. Nobody was really sure exactly what it was doing there. We think it’s playing an important catalytic role.”
DNA polymerase adds the nucleotides guanine, adenine, thymine, cytosine (G-A-T-C) to DNA by removing a proton from the end of the growing strand through reaction with a water molecule. “When we say in the study that a water molecule serves as the base, it serves as a base to remove a proton, an acid base chemistry. What’s left there after you remove the proton is much more chemically active to react with a new nucleotide that needs to be added to the DNA,” Roston said.
The chemistry needs multiple proton transfers in a complex active site. Experimental probes using X-ray crystallography have been unable to distinguish among the many possible reaction pathways.
“Simulations offer a complement to crystallography because you can model in all the hydrogens and run molecular dynamics simulations, where you allow all the atoms to move around in the simulation and see where they want to go, and what interactions are helping them get where they need to go,” Roston said. “Our role was to do these molecular dynamics simulations and test different models for how the atoms are moving around during the reaction and test different interactions that are helping that along.”
The number of energy calculations needed to complete the molecular dynamics simulations was huge, on the order of 10e8 to 10e9 for the system with thousands of atoms and many complex interactions. That’s because timesteps at the right resolution are on the order of femtoseconds, 10e-15 seconds.
“Chemical reactions, life, doesn’t happen that quickly,” Roston said. “It happens on a timescale of people talking to each other. Bridging this gap in timescale of many, many orders of magnitude requires many steps in your simulations. It very quickly becomes computationally intractable.”
“One of the great things about XSEDE is that we can take advantage of a ton of computational power,” Roston added. Through XSEDE, Roston and his colleagues used about 500,000 CPU hours on the Comet system at the San Diego Supercomputer Center. Comet allowed them to simultaneously run many different simulations that all feed off one another.
Said Roston: “DNA replication is what life is about. We’re getting at the heart of how that happens, the really fundamental process to life as we know it on Earth. This is so important, we should really understand how it works at a deep level. But then, there are also important aspects of technology, such as CRISPR, that take advantage of this kind of work to develop systems to manipulate DNA. Understanding the details of how life has evolved to manipulate DNA will play a role in feeding our understanding and our ability to harness technologies in the future.”
‘Molecular simulation of mechanical properties and membrane activities of the ESCRT-III complexes’ was published online in the journal Biophysical Journal in February 2020. The study co-authors are Taraknath Mandal and Qiang Cui of Boston University; Wilson Lough, Saverio E. Spagnolie, and Anjon Audhya of the University of Wisconsin-Madison. Study funding came from the National Science Foundation. Computations are also supported in part by the Shared Computing Cluster, which is administered by Boston University’s Research Computing Services.
‘Extensive free-energy simulations identify water as the base in nucleotide addition by DNA polymerase’ was published in the Proceedings of the National Academy of Sciences in December 2019. The study co-authors are Daniel Roston of the University of California San Diego; Darren Demapan of the University of Wisconsin-Madison; and Qiang Cui of Boston University. Study funding came from the National Institutes of Health.
###
Media Contact
Jorge Salazar
[email protected]
512-471-3980
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}