XSEDE’s Stampede2, bridges speed skin cancer research
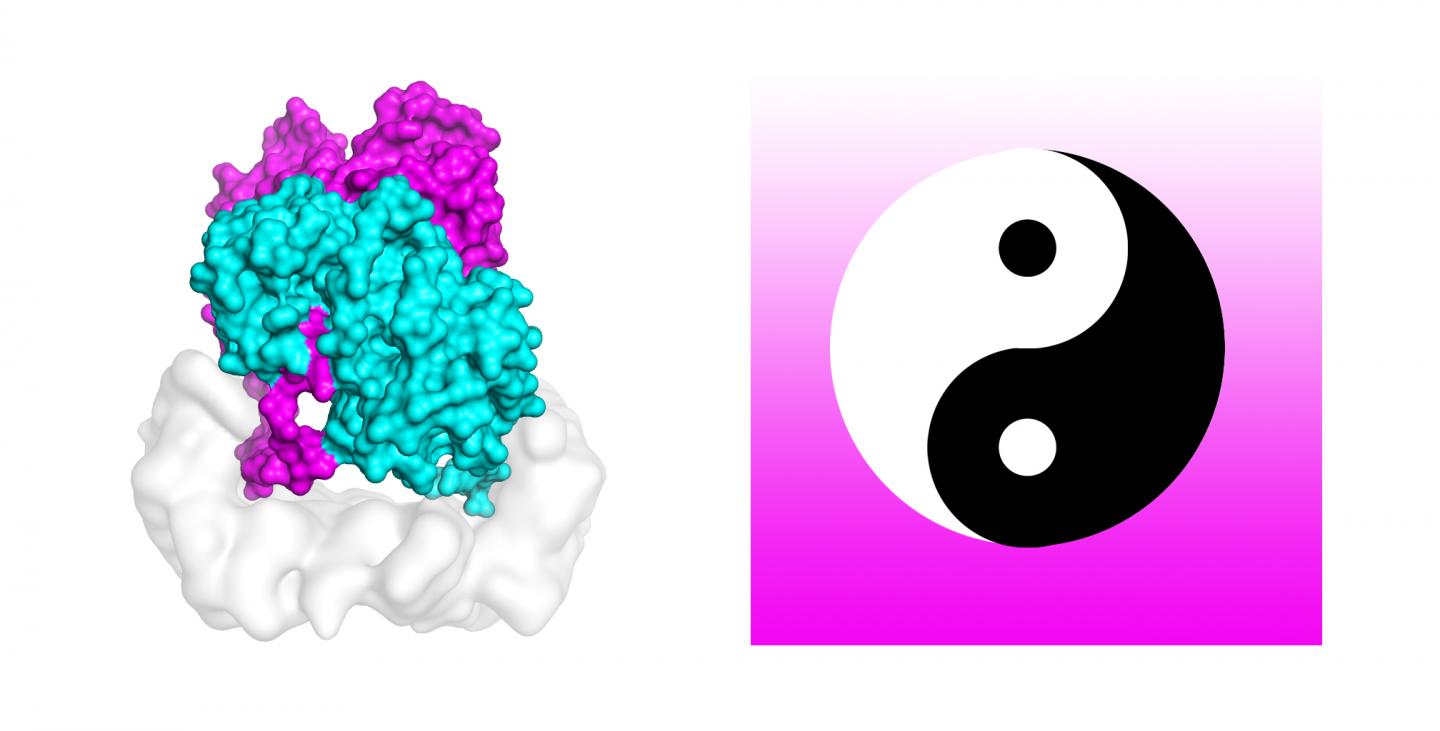
Credit: Karandur et al., TACC
It starts off small, just a skin blemish. The most common moles stay just that way — harmless clusters of skin cells called melanocytes, which give us pigment. In rare cases, what begins as a mole can turn into melanoma, the most serious type of human skin cancer because it can spread throughout the body.
Scientists are using powerful supercomputers to uncover the mechanism that activates cell mutations found in about 50 percent of melanomas. The scientists say they’re hopeful their study can help lead to a better understanding of skin cancer and to the design of better drugs.
In 2002, scientists found a link between skin cancer and mutations of B-Raf (Rapidly Accelerated Fibrosarcoma) kinase, a protein that’s part of the signal chain that starts outside the cell and goes inside to direct cell growth. This signal pathway, called the Ras/Raf/Mek/Erk kinase pathway, is important for cancer research, which seeks to understand out of control cell growth. According to the study, about 50 percent of melanomas have a specific single mutation on B-Raf, known as the valine 600 residue to glutamate (V600E).
B-Raf V600E thus became an important drug target, and specific inhibitors of the mutant were developed in the following years. The drugs inhibited the mutant, but something strange happened. Paradoxically, quieting the mutant had a down side. It activated the un-mutated, wild-type B-Raf protein kinases, which again triggered melanoma.
“With this background, we worked on studying the structure of this important protein, B-Raf,” said Yasushi Kondo, a postdoctoral researcher in the John Kuriyan Lab at UC Berkeley. Kondo is the co-author of an October 2019 study in the journal Science that determined the structure of the complex of proteins that make up B-Raf and also found how the paradoxical B-Raf activation happens.
“We aimed to study the more native-like state of the protein to understand how it’s regulated in the cells, because most of the studies have been focused on the isolated kinase domain and how the drugs bind to the kinase domain.” Kondo said.
The full-length B-Raf protein is made of several domains linked by disordered regions, something too unwieldy for scientists to yet image. Kondo’s technique was to use intein chemistry to make smaller fragments, then stitch them up to get the full structure.
“As a result, we obtained an active form of the full-length B-Raf dimer called B-Raf co-purified with 14-3-3 dimer, a scaffolding protein bound to the phosphorylated B-Raf C-terminal tail,” Kondo said.
Kondo’s group used cryo-electron microscopy (cryo-EM) to determine the structure of B-Raf 14-3-3 complex, basically cryogenically freezing the protein complex, which kept it in a chemically-active, near-natural environment. Next they flashed it with electron beams to obtain thousands of ‘freeze frames.’ They sifted out background noise and reconstructed three-dimensional density maps that showed previously unknown details in the shape of the molecule. And for proteins, form follows function.
Kondo explained that the structure revealed an asymmetric organization of the complex, formed by two sets of internally symmetrical dimers, or pairs of bonded molecules. “We propose that this unexpected arrangement enables asymmetric activation of the B-Raf dimer, which is a mechanism that provides an explanation of the origin of the paradoxical activation of B-Raf by small molecule inhibitors,” Kondo said.
Detailed analysis of the asymmetrical B-Raf 14-3-3 complex structure showed another unexpected structural feature, described as the distal tail segment, DTS for short, of one B-Raf molecules. Kondo said the tail of one is bound to the active site of the other, blocking its activity by competing with ATP binding. The blocked B-Raf molecule is stabilized in the active conformation. “We interpreted this structure that this blocked B-Raf molecule functions as an activator and stabilizes the other B-Raf receiver through the dimer interface,” Kondo said.
Curiously enough, the authors compare the B-Raf dimer to the Chinese yin-yang circular symbol of interconnected opposites joined at the tail. “From looking at the subject, it’s very clear that one is not capable of phosphorylating the downstream molecule, which is necessary for cell growth. The other molecule is clearly the one to do the job. In this set of two molecules, we clearly see one is doing the supporting job, and the other one is doing the actual work. It really does look like Yin and Yang in this B-Raf 14-3-3 complex we solved,” Kondo said.
Looks, though, can be deceiving. Scientists used computer simulations to help verify that they were really onto something. “We ran molecular dynamics simulations of this complex of the B-Raf dimer bound to a 14-3-3 dimer to test the stability of the asymmetric conformation,” said study co-author Deepti Karandur, also a postdoctoral researcher at the John Kuriyan Lab of UC Berkeley; she’s also a postdoctoral fellow at the Howard Hughes Medical Institute. “We didn’t know why the conformation was asymmetric, or what role it played in maintaining the active state of the enzyme,” Karandur said.
They started the simulations using the structure that Kondo had solved by cryo-EM, with the DTS segment running from one kinase into the active site of the other. Then they ran a second set of simulations with the DTS segment removed.
“What we found was that in the system without the distal tail segment, the entire complex is not stable,” Karandur explained. “The kinase domains move with respect to the scaffolding, the 14-3-3 dimer. In one of our simulations, the dimer state of B-Raf itself, which experiments have shown is necessary to maintain the active state of this kinase, it fell apart, indicating that this distal tail segment, DTS, is necessary to actually maintain this complex in this asymmetric conformation, which in turn is necessary to maintain the kinase dimer in the stable asymmetric dimer active state.”
One of the main results of the study was finding the mechanism of action that switches on the B-Raf kinase complex of two B-Raf kinases and two 14-3-3 scaffolding proteins, where on B-Raf kinase is the activator, and the other is the receiver.
“The tail of the receiver molecule is inside the active site of the activator, so the activator cannot work as an enzyme,” Kondo said. “Instead, the activator molecule stabilizes the active conformation of the receiver molecule. The 14-3-3 scaffold protein facilitates this arrangement, so that the tail insertion only happens to one kinase molecule. We hypothesize that when there is no 14-3-3 binding, both kinases can be blocked by the insertion of the DTS, but this needs to be tested.”
The study’s computational challenges involved molecular dynamics simulations that modeled the protein at the atomic level, determining the forces of every atom on every other atom for a system of about 200,000 atoms at time steps of two femtoseconds.
“For small systems, we can see what’s happening relatively quickly, but for large systems like these, especially large biomolecular systems, these changes happen on like nanosecond timescales, microsecond timescales, or even millisecond timescales,” Karandur said.
Karandur and colleagues turned to XSEDE, the NSF-funded Extreme Science and Engineering Discovery Environment, for allocation time on the Stampede2 supercomputer at the Texas Advanced Computing Center (TACC) to do the simulations, as well as the Bridges system at the Pittsburgh Supercomputer Center to investigate other proteins in the pathway. Stampede2’s Skylake processor nodes, networked with Intel Omnipath, made quick work of the optimized-for-supercomputers NAMD molecular dynamics simulations.
“Stampede2 runs very, very fast, and it’s very efficient. We generated a total of about 1.5 microseconds of trajectories for our systems in about four to six weeks. Whereas, if we ran it on our own in-house cluster it would have taken us months or longer,” Karandur said.
About XSEDE, Karandur commented: ” I think it’s an amazing resource. I’ve been running simulations starting from when I was a graduate student. XSEDE made it possible for us to access timescales that are biologically relevant. Everything that happens in a cell, happens on microsecond timescales, to millisecond timescales, to longer. When I was starting, we could not run this simulation on any system anywhere. I mean, it would have taken five years, or more. To be able to do it in weeks and say, okay, we know understand why this is important so we can now start to gain real understanding into how the biology happens, is just amazing,” Karandur said.
And there remains a lot to be discovered about B-Raf. It’s just one link in the signal chain that governs cell growth and cancer.
“The structure that was resolved in this paper is part of a large, multi-domain system,” Karandur explained. “We don’t know what this complete protein looks like. We don’t see it in the structure. We don’t know what its dynamics look like, and how all these other parts of the protein play a role in maintaining the active state, or converting it from the inactive state to the active state.”
She furthered that as the system gets bigger, the pertinent structural changes happen over longer timescales, and bigger supercomputers are needed to handle the complexity, such as the NSF-funded Frontera supercomputer, also at TACC.
“Frontera is getting there. We’re very excited about this. We are in the process of getting an allocation on Frontera,” Karandur said.
For non-scientists, this fundamental research could yield insight leading to better drugs for skin cancer.
“The paradoxical activation of Raf kinase by these B-Raf-specific inhibitors turn normal cells to tumors during skin cancer treatment,” Kondo said. Understanding the mechanism of this phenomenon will allow us to design better drugs. Hopefully, our study can contribute to the understanding of this step. In addition, we found mutations in this link between the Kinase domain and the 14-3-3 binding element of the B-Raf molecule, which was never shown before. This mutation reduces the activity of B-Raf in the cells. It’s also indicating that this part of the kinase domain can be a target point to develop new kinds of B-Raf inhibitors.”
Said Karandur: “There’s a lot of dynamics happening in the cell. We are, largely because of XSEDE, only starting to be able to look at things like that. Going forward, the only way we can continue to look at things is by using very, very large supercomputers, because the calculations require a lot of computational power. It’s really exciting to be able to actually see these things happen and to say, here are how things change at the atomic level; here are these interactions between these two atoms form or break, and that translates into this huge change at the global level in the overall structure of the protein, and how it interacts with other proteins, or other molecules in the cell. We’re very excited about where it will go in the future.”
###
The study, “Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases,” was published Oct. 4, 2019 in the journal Science. The authors are Yasushi Kondo, Deepti Karandur, John Kuriyan, and Kathryn Wong of UC Berkeley; Jana Ognjenovic? and Alan Merk of the National Cancer Institute; Saikat Banerjee, Kayla Kulhanek, Jeroen P. Roose of UC San Francisco; and Sriram Subramaniam of the University of British Columbia, Vancouver. Study funding came from the National Institutes of Health, the Aqueduct Foundation, the VGH & UBC Hospital Foundation, and the Government of Canada.
Stampede2 and Bridges are allocated resources of the Extreme Science and Engineering Discovery Environment (XSEDE) funded by the National Science Foundation (NSF).
Media Contact
Jorge Salazar
[email protected]
512-471-3980
Original Source
https:/
Related Journal Article
http://dx.


{kind=link}