Osaka City University breaks from convention with a true quantum algorithm that can calculate energy differences between the electronic ground and excited spin states of molecular systems
Credit: K. Sugisaki, K. Sato and T. Takui.
Understanding how the natural world works enables us to mimic it for the benefit of humankind. Think of how much we rely on batteries. At the core is understanding molecular structures and the behavior of electrons within them. Calculating the energy differences between a molecule’s electronic ground and excited spin states helps us understand how to better use that molecule in a variety of chemical, biomedical and industrial applications. We have made much progress in molecules with closed-shell systems, in which electrons are paired up and stable. Open-shell systems, on the other hand, are less stable and their underlying electronic behavior is complex, and thus more difficult to understand. They have unpaired electrons in their ground state, which cause their energy to vary due to the intrinsic nature of electron spins, and makes measurements difficult, especially as the molecules increase in size and complexity. Although such molecules are abundant in nature, there is a lack of algorithms that can handle this complexity. One hurdle has been dealing with what is called the exponential explosion of computational time. Using a conventional computer to calculate how the unpaired spins influence the energy of an open-shell molecule would take hundreds of millions of years, time humans do not have.
Quantum computers are in development to help reduce this to what is called “polynomial time”. However, the process scientists have been using to calculate the energy differences of open-shell molecules has essentially been the same for both conventional and quantum computers. This hampers the practical use of quantum computing in chemical and industrial applications.
“Approaches that invoke true quantum algorithms help us treat open-shell systems much more efficiently than by utilizing classical computers”, state Kenji Sugisaki and Takeji Takui from Osaka City University. With their colleagues, they developed a quantum algorithm executable on quantum computers, which can, for the first time, accurately calculate energy differences between the electronic ground and excited spin states of open-shell molecular systems. Their findings were published in the journal Chemical Science on 24 Dec 2020.
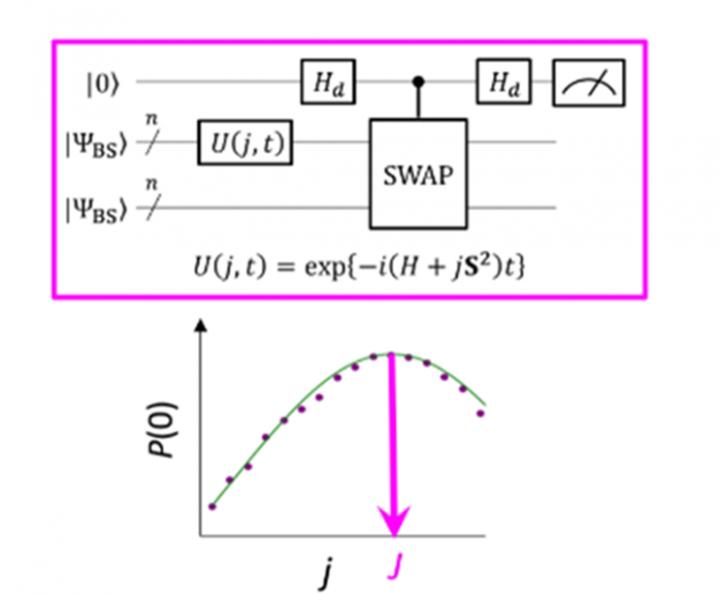
The energy difference between molecular spin states is characterized by the value of the exchange interaction parameter J. Conventional quantum algorithms have been able to accurately calculate energies for closed-shell molecules “but they have not been able to handle systems with a strong multi-configurational character”, states the group. Until now, scientists have assumed that to obtain the parameter J one must first calculate the total energy of each spin state. In open-shell molecules this is difficult because the total energy of each spin state varies greatly as the molecule changes in activity and size. However, “the energy difference itself is not greatly dependent on the system size”, notes the research team. This led them to create an algorithm with calculations that focused on the spin difference, not the individual spin states. Creating such an algorithm required that they let go of assumptions developed from years of using conventional computers and focus on the unique characteristics of quantum computing – namely “quantum superposition states”.
“Superposition” lets algorithms represent two variables at once, which then allows scientists to focus on the relationship between these variables without any need to determine their individual states first. The research team used something called a broken-symmetry wave function as a superposition of wave functions with different spin states and rewrote it into the Hamiltonian equation for the parameter J. By running this new quantum circuit, the team was able to focus on deviations from their target and by applying Bayesian inference, a machine learning technique, they brought these deviations in to determine the exchange interaction parameter J. “Numerical simulations based on this method were performed for the covalent dissociation of molecular hydrogen (H2), the triple bond dissociation of molecular nitrogen (N2), and the ground states of C, O, Si atoms and NH, OH+, CH2, NF and O2 molecules with an error of less than 1 kcal/mol”, adds the research team.
“We plan on installing our Bayesian eXchange coupling parameter calculator with Broken-symmetry wave functions (BxB) software on near-term quantum computers equipped with noisy (no quantum error correction) intermediate-scale (several hundreds of qubits) quantum devices (NISQ devices), testing the usefulness for quantum chemical calculations of actual sizable molecular systems.”
###
We are Osaka City University – the oldest research university in Osaka. With 9 undergraduate faculties and 11 graduate schools all dedicated to making urban life better, energy cleaner, and people healthier and happier, we have won numerous awards and have produced 2 Nobel laureates. For more information, please visit our website at https:/
Media Contact
James Gracey
[email protected]
Original Source
https:/
Related Journal Article
http://dx.



{kind=link}