Credit: University of Pennsylvania
Mitochondria, the mighty energy factories of the cell, often malfunction in cancer, as well as in other conditions such as aging, neurodegenerative disease and heart disease. Whether these changes in mitochondria actually contribute to the spread of cancer, however, has been controversial.
In a new report published in the journal Cell Discovery, a team led by researchers in the University of Pennsylvania School of Veterinary Medicine has identified a mechanism by which mitochondria can drive changes in nuclear gene expression that are associated with tumor progression. The epigenetic process is carried out by a protein that is triggered in response to mitochondrial oxidative or metabolic stress. When this interaction was blocked by chemical compounds, the team was able to reduce cancer gene expression.
The work was led by Penn Vet's Manti Guha, a research assistant professor, and Narayan Avadhani, a professor.
"Our study provides a rigorous demonstration of a link between mitochondrial function and nuclear gene expression," said Avadhani. "Since we know that this type of signaling has a direct role in the early stages of cancer progression, the protein involved could be a very valuable target for alleviating this signaling and possibly cancer progression."
Avadhani's group has published several papers with evidence that depletions and disruptions in the number and function of mitochondria contribute to cancer, zeroing in on the idea that mitochondria trigger a stress signal that alerts the nucleus that something is amiss.
This new work drills in on the specifics of this "SOS" alert. The researchers turned their attention to the protein hnRNPA2, short for heterogenous ribonucleoprotein A2. They had identified this protein years earlier as one that was activated in response to mitochondrial stress and also bound tightly to nuclear DNA. This protein had previously been known as an RNA binding protein but had also been shown to play a role in driving cancer progression in addition to associating with the DNA of telomeres, the "caps" of chromosomes.
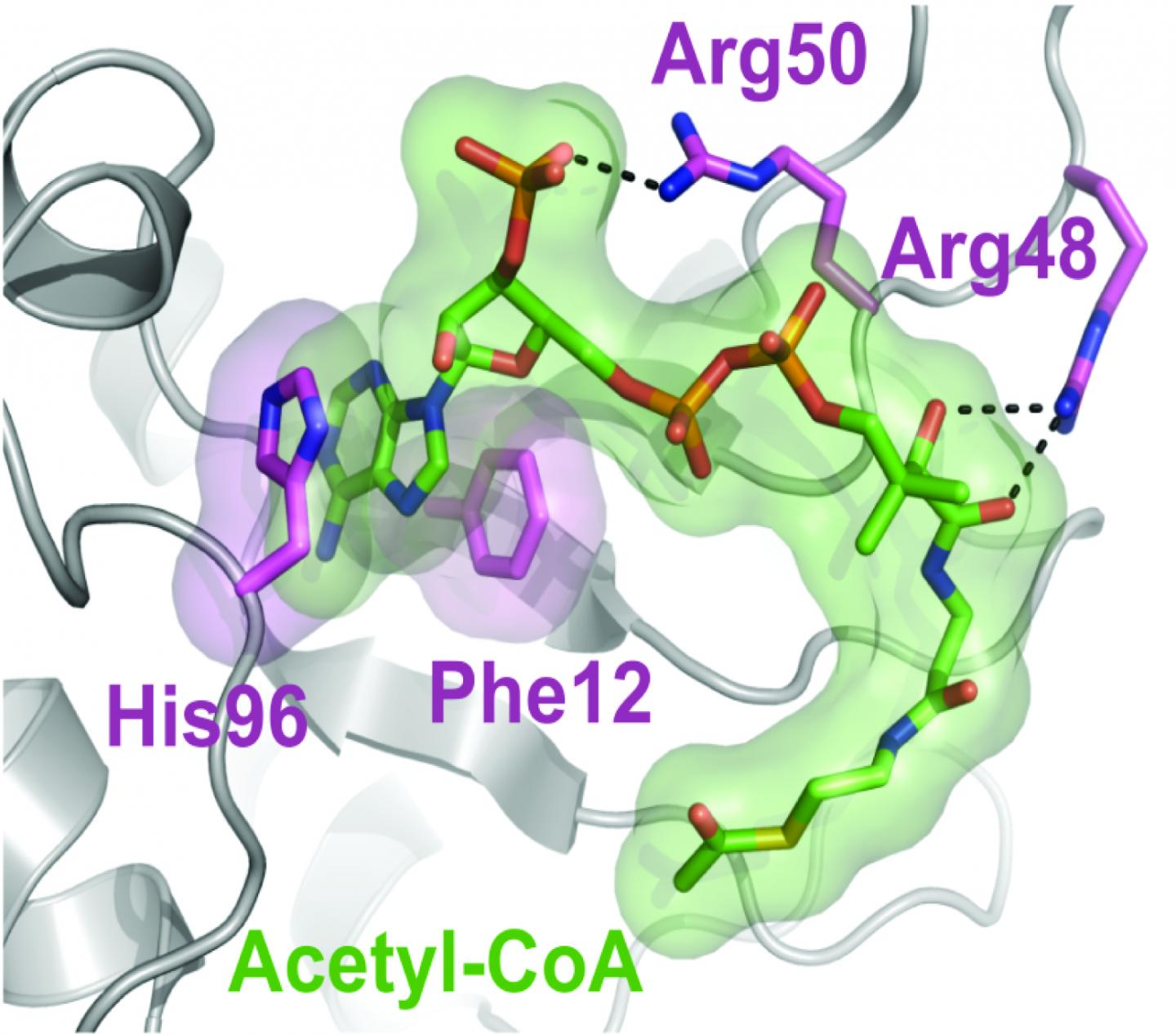
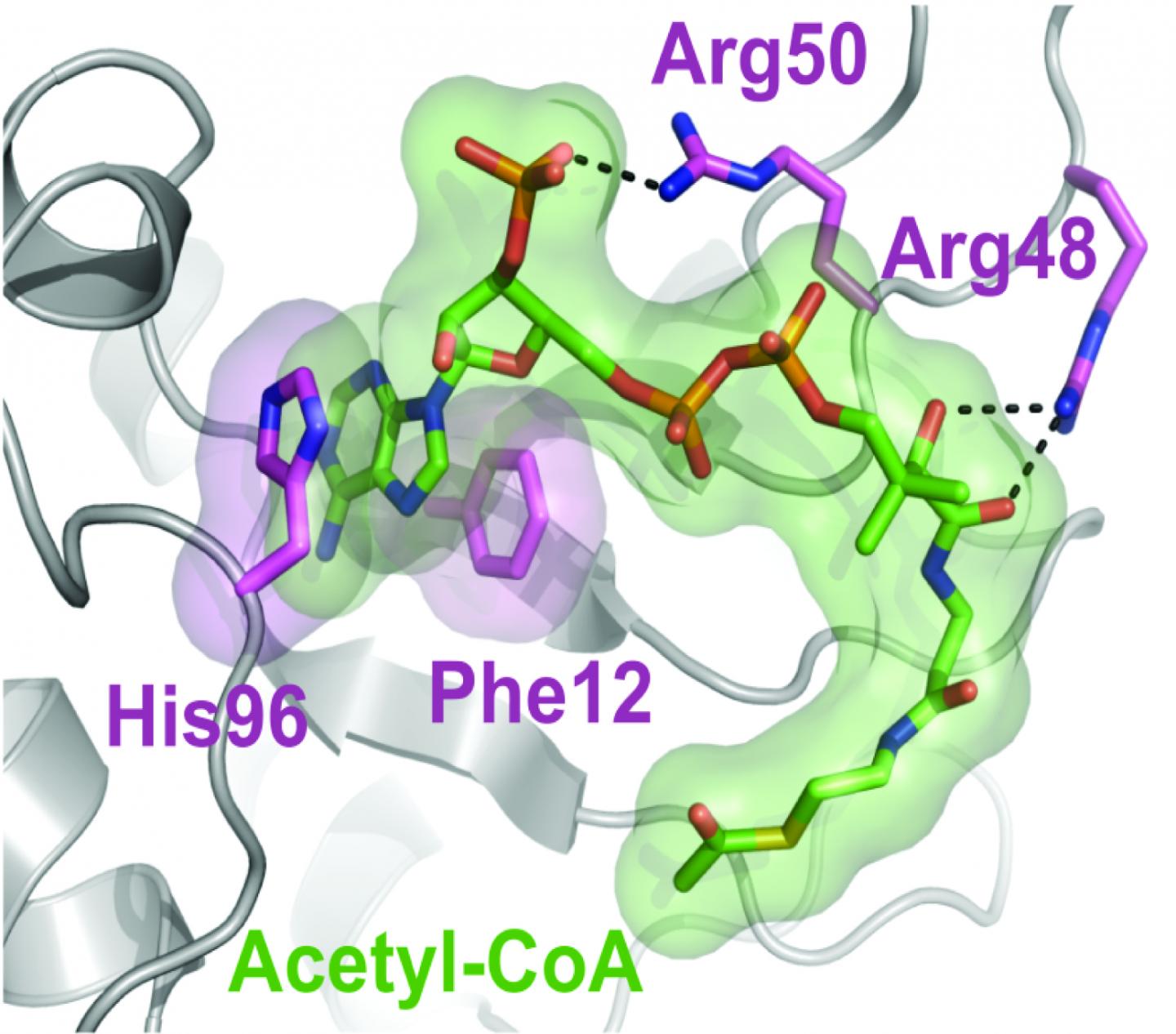
The researchers evaluated the role of hnRNPA2 in the mitochondria-to-nucleus communication by manipulating its activity in cells with depleted levels of mitochondria. They found that hnRNPA2 activated gene promoters of stress-associated genes in the nucleus by binding to them. A closer examination revealed that hnRNPA2 turned on the genes by modifying a histone, tagging it with acetyl-CoA groups. This modification, known as acetylation, helps open up the tightly packed chromatin, allowing DNA transcription to proceed more easily.
In addition to these stress-associated genes, which cancer cells activate in order to maintain growth and achieve immortality, tumorigenic cells also activate telomerase, an enzyme that ensures that the cells' telomeres are kept at an appropriate length to avoid sending the cells into dormancy. The researchers again tested hnRNPA2's role in this process, finding that while telomerase levels increased in mitochondria-depleted cells, blocking hnRNPA2 reversed this effect.
Further investigation allowed the team to confirm that the protein acted as a catalyst to transfer acetyl-CoA groups onto proteins. They also developed a structural model of hnRNPA2 that enabled them to identify the physical sites the protein where acetyl-CoA binding occurs. When the researchers produced versions of hnRNPA2 with those sites mutated, the protein's acetyltransferase activity was greatly reduced.
As a final test of the link between hnRNPA2 and tumor progression, the researchers showed that, though cells with reduced levels of mitochondrial DNA had an increased ability to invade, as tumor cells do, cells expressing mutant hnRNPA2 lacked this ability.
"Now we have not only identified the protein but the actual sites on that protein that, if we mutate, we can stop that modification," said Guha. "Just changing the protein on those two residues seems to actually reverse the entire process that drives tumorigenesis."
Looking ahead, the researchers hope to test small molecules that block the catalytic activity of hnRNP2 to see if they can prevent the epigenetic activity that leads to cells taking on invasive properties.
###
Study coauthors included Penn Vet's Satish Srinivasan, J-K Fang and Gordon Ruthel; Penn Perelman School of Medicine's F. Brad Johnson, M. Rebecca Glineburg and Hiroshi Nakagawa; Stony Brook University's Kip Guja, Edison Mejia and Miguel García-Díaz; the University of Pittsburgh's Brett A. Kaufman; the Children's Hospital of Philadelphia's Eric F. Rappaport; Temple University's Andres Klein Szanto; and Jeelan Basha and Tapas Kundu of the Jawaharlal Nehru Centre for Advanced Scientific Research.
The study was supported by the National Institutes of Health, the Harriet Ellison Woodward Trust and Mitochondria Research Affinity Core grants.
Media Contact
Katherine Unger Baillie
[email protected]
215-704-7540
@Penn
http://www.upenn.edu/pennnews
############
Story Source: Materials provided by Scienmag


{kind=link}