Credit: UC San Diego Health Sciences
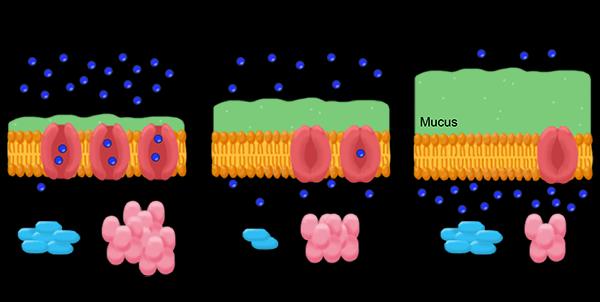
Cystic fibrosis is caused by an inherited mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Due to this mutation, the CFTR protein doesn’t embed in cell membranes to form a channel for chlorine ions the way it should. As a result, mucus-producing cells secrete a thicker-than-normal mucus that can create blockages in the lungs and digestive system. In the lungs, this thicker mucus can help bacteria thrive, making lung infections a serious and chronic problem for many people living with cystic fibrosis.
Yet some people with cystic fibrosis don’t develop lung infections as early or as frequently as others. Researchers at University of California San Diego School of Medicine recently discovered that genetic variations dampening expression of another gene, called RNF5, offer a likely explanation.
In the study, published December 10, 2019 in eLife, the team found that individuals with cystic fibrosis who carry specific genetic variants lowering expression of RNF5 have more mutant CFTR protein on their cell surfaces. Even if the CFTR protein isn’t totally functional, it’s likely better than having none at all, the researchers said.
Cystic fibrosis symptoms can be significantly improved with relatively new medications that boost CTFR function. But there are two caveats: The drugs are extremely expensive and they don’t work for everyone.
“The cystic fibrosis field is trying to figure out what are the modifiers across the genome that increase or decrease the probability that an individual patient will respond to these expensive drugs,” said senior author Kelly A. Frazer, PhD, professor of pediatrics and director of the Institute for Genomic Medicine at UC San Diego School of Medicine. “RNF5 may be one of these modifier genes.”
The RNF5 gene is located in the Major Histocompatibility Complex (MHC). Genes in this region encode molecules that are displayed on the surface of most cells in the body. They play an important role in how the body responds to infections. Scientists have long known that everyone has their own set of MHC gene variations, and that they make people more or less susceptible to infections or autoimmune diseases. But because the genes present in the MHC region are so dense with variations, they have not been well-studied for their direct link to diseases.
Frazer and colleagues took a new approach to analyzing MHC gene variants by grouping them. That allowed the team to more easily identify associations between genetic variation, gene expression levels and their effects on complex diseases. The researchers applied this approach with whole genome sequencing of induced pluripotent stem cells derived from 419 people. That translated into investigating more than 4,000 traits, from which they identified 180 associated with variants in the MHC region.
“We’ve known there’s an association between MHC genes and bacterial colonization in patients with cystic fibrosis, but no one knew why,” Frazer said. “We assumed it was due to MHC’s involvement in the immune system. But now we know that’s likely not the only mechanism — different expression levels of RNF5 may also play an important role.”
“This study uncovered a new aspect of cystic fibrosis — one that could lead to new drug design and development, and allow clinicians to better tailor treatments,” said first author Matteo D’Antonio, PhD, a project scientist in Frazer’s lab. “But that’s just one example of how we might employ this new genetic analysis technique. Now we have a road map, and we can apply it to other diseases and cell types.”
###
Additional co-authors include: Joaquin Reyna, David Jakubosky, Margaret K.R. Donovan, Hiroko Matsui, Naoki Nariai, Agnieszka D’Antonio-Chronowska, UC San Diego; and Marc-Jan Bonder, Oliver Stegle, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Trust Genome Campus.
Media Contact
Heather Buschman
[email protected]
858-249-0456
Related Journal Article
http://dx.


{kind=link}