Credit: JING Gongchao
A new algorithm may reduce the need for expensive, time-consuming whole-genome sequencing computations to understand how a microbiome functions. A team led by JING Gongchao of the Qingdao Institute of BioEnergy and Bioprocess Technology (QIBEBT) of the Chinese Academy of Sciences (CAS) and SU Xiaoquan of Qingdao University, published their approach, called Meta-Apo, on Jan. 6 in BMC Genomics.
Researchers routinely sequence samples of microbial communities found on human skin, in human guts, and in the environment to understand what genes they contain with the ultimate goal of understanding how they function.
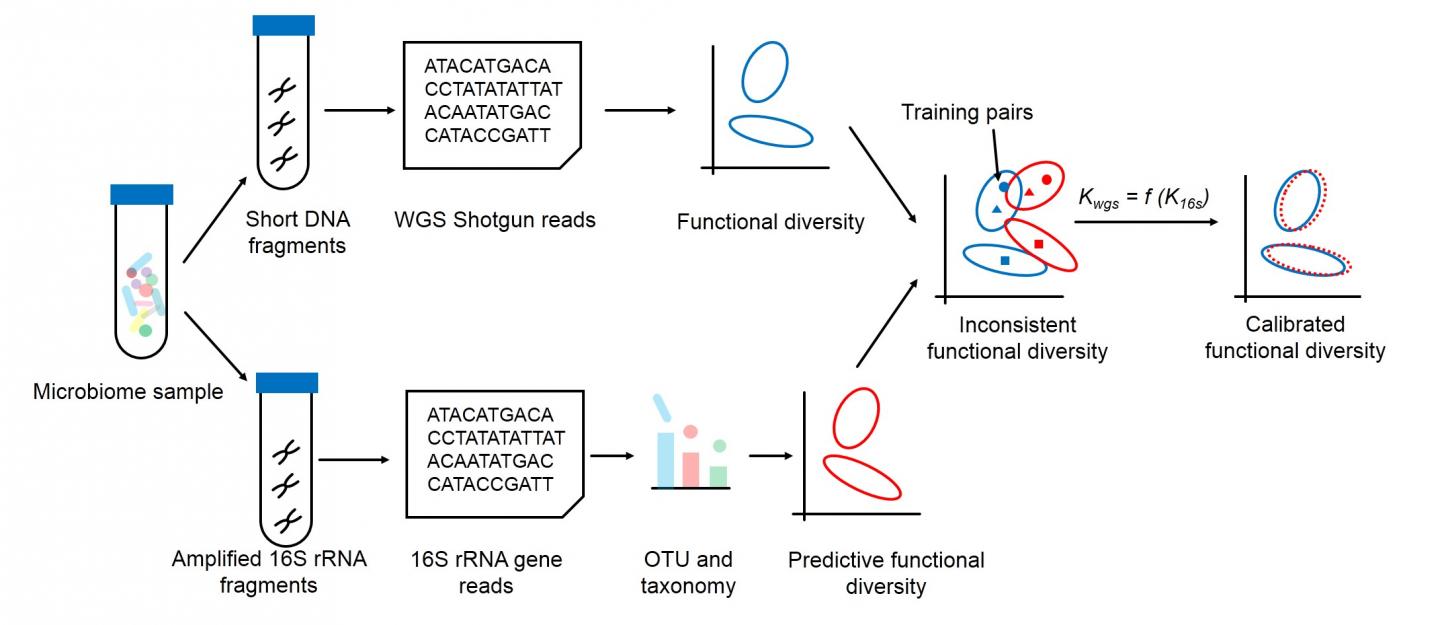
According to JING, the first author of the study, two main approaches exist: shotgun whole-genome sequencing and 16S rRNA gene amplicons. Whole-genome sequencing requires significant sequencing cost as well as computing power to determine all of the genes and their functions in a single sample, while 16S rRNA gene amplicons can quickly tease out a sample’s specific gene for taxonomy information and thus predict how they function.
“However, due to the potential biases in how the amplicons are prepared and gene profile variation among genomes, functional profiles predicted from 16S amplicons may deviate from whole-genome sequencing ones, resulting in misleading results,” said JING. “Our approach, Meta-Apo, greatly reduces or even eliminates such deviation, deducing more consistent diversity patterns between the two approaches.”
Meta-Apo matches pairs of data from whole-genome sequencing and 16S amplicons – each pair is sequenced via both methods – to teach new 16S amplicon samples to better recognize gene function. The results are much more consistent with the whole-genome sequencing results.
“Tests of Meta-Apo on more than 5,000 16S amplicon human microbiome samples from four body sites showed the deviation between the two strategies is significantly reduced by using only 15 training sample pairs,” JING added. “Moreover, Meta-Apo enables cross-platform functional comparison between whole-genome sequencing and amplicon samples, greatly improving 16S-based microbiome diagnoses.”
To test this experimentally, the researchers were able to improve the accuracy of a gingivitis diagnosis from 65% to 95% percent using the 16S-derived functional profiles, produced by training the whole-genome sequencing pairs.
“With the low cost of 16S-amplicon sequencing, Meta-Apo can produce a reliable, high-resolution view of microbiome function equivalent to that offered by shotgun whole-genome sequencing,” SU, senior author of the study, explained.
###
Other contributors include LIU Lu and XU Jian, Single-Cell Center, CAS Key Laboratory of Biofuels, Qingdao Institute of BioEnergy and Bioprocess Technology (QIBEBT); CUI Wenzhi, College of Control Science and Engineering, China University of Petroleum; and ZHANG Yufeng, College of Computer Science and Technology, Qingdao University.
The Natural Science Foundation of China, the Shandong Provincial Natural Science Foundation and China Postdoctoral Science Foundation supported this work.
Media Contact
CHENG Jing
[email protected]
Original Source
https:/
Related Journal Article
http://dx.


{kind=link}