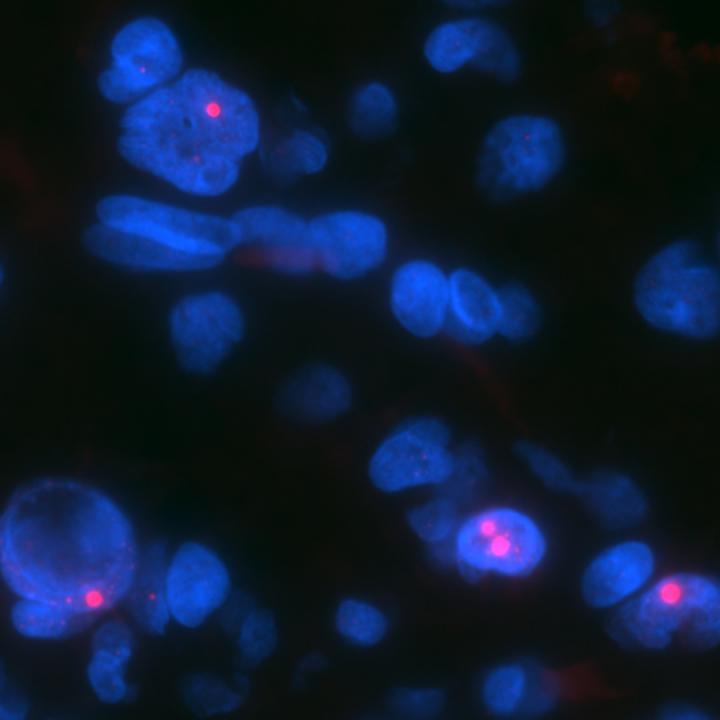
Credit: Chris Heaphy
Two recently discovered genetic differences between brain cancer cells and normal tissue cells — an altered gene and a snippet of noncoding genetic material — could offer clues to tumor behavior and potential new targets for therapy, Johns Hopkins scientists report.
In a report on one of the two findings, published online Sept. 20 in Acta Neuropathologica, the investigators identified alterations in a protein known as ATRX in human brain tumors that arise as part of a genetically inherited condition known as neurofibromatosis type 1 (NF1). The disorder, marked initially by benign tumors on nerves, often leads to brain cancer, and although most NF1-related malignancies are nonaggressive, a fraction are "high-grade" and difficult to treat, experts say.
Study leader Fausto J. Rodriguez, M.D., associate professor of pathology at the Johns Hopkins University School of Medicine and member of the Johns Hopkins Kimmel Cancer Center, says the new study sought to sort out what makes the more aggressive NF1-related tumors genetically different from low grade tumors and normal, healthy cells.
Research from other scientists at Johns Hopkins, he says, had suggested that some tumors, particularly those that affect the nervous system, have mutations in the ATRX gene, which produces proteins that appear to maintain the length of telomeres, repetitive segments of DNA on the ends of chromosomes that typically shorten each time a cell divides. Telomere shortening limits the number of divisions that cells can undergo. By keeping telomeres long, ATRX mutations give cells the ability to endlessly divide, a hallmark of cancer.
To see what role ATRX mutations might play in NF1-related brain cancers, Rodriguez and his colleagues examined samples of both high- and low-grade gliomas — tumors that arise from parent cells of nerve-supporting cells called glia — which were removed by surgery from 27 patients with NF1.
They tested the samples for longer telomeres that weren't the product of a protein called telomerase, which helps maintain telomere length in healthy cells and more commonly in other cancers. This nontelomerase-associated telomere lengthening is known as alternative lengthening of telomeres or ALT. They also tested for loss of the protein product of ATRX caused by mutations in this gene.
About a third of the 27 samples had ALT and loss of the ATRX protein. The presence of both factors occurred in seven of 12 patients with high-grade tumors but only two of 14 with low-grade cancers.
If further studies confirm the role of the mutation, Rodriguez says, researchers might be able to develop anticancer agents to target cells with ATRX mutations or ALT to limit telomere length. In the meantime, he adds, gaining a better understanding of aggressive NF1-related tumors allows researchers to develop more realistic models of these cancers in the lab.
In a second study, described online Oct. 14 in Modern Pathology, the Johns Hopkins investigators sought a genetic source that could accurately identify subsets of low-grade pediatric gliomas, the most frequent tumors of the central nervous system in children. Unlike many other types of cancerous tumors, low-grade pediatric gliomas appear to have few genetic mutations, leaving the biology of these cancers a mystery, says Rodriguez.
In an effort to unravel that mystery, Rodriguez and his colleagues focused on the ways those cancer cells regulate the expression of genes or whether they make their constituent proteins. One way to control expression, he explains, is through microRNA, small pieces of noncoding genetic material that control whether a gene's protein is generated from the DNA blueprints inside cell nuclei.
The researchers analyzed the amounts of 800 different microRNAs in nine different types of low-grade gliomas or related cancers. Of all of these, one in particular caught the researchers' eyes: a microRNA called miR-487b, which was consistently underexpressed in tumor tissue compared to healthy cells.
To pin down the role of this microRNA, the researchers artificially increased its levels in lines of laboratory-grown human cancer cells that appeared to make too little of it, in turn bringing down the levels of the protein it affects. Unlike unaltered cancer cells, these new cells formed 30 percent fewer colonies and had decreased levels of other proteins, such as Nestin, by a third. Nestin is known to be important in both early development and in cancers.
Eventually, Rodriguez says, physicians might be able to look at the levels of this and other microRNAs in blood or cerebrospinal fluid to test for the presence of cancer. Researchers might also be able to target microRNAs directly, altering their levels to make cancer cells less likely to form tumors.
"By gaining a better understanding of the fine genetic differences between cancers and healthy tissues, we can develop better therapeutic or prognostic strategies," he says.
###
Funding for the studies was provided by the Childhood Brain Tumor Foundation, the Pilocytic/Pilomyxoid Astrocytoma Fund, and Lauren's First and Goal.
Other Johns Hopkins researchers who participated in the Acta Neuropathologica study include Maria Adelita Vizcaino, Jaishri Blakeley and Christopher M. Heaphy. Other Johns Hopkins researchers who participated in the Modern Pathology study include Heather Marion Ames, Ming Yuan, Maria Adelita Vizcaino and Wayne Yu.
Media Contact
Vanessa Wasta
[email protected]
410-614-2916
@HopkinsMedicine
http://www.hopkinsmedicine.org
############
Story Source: Materials provided by Scienmag

{kind=link}