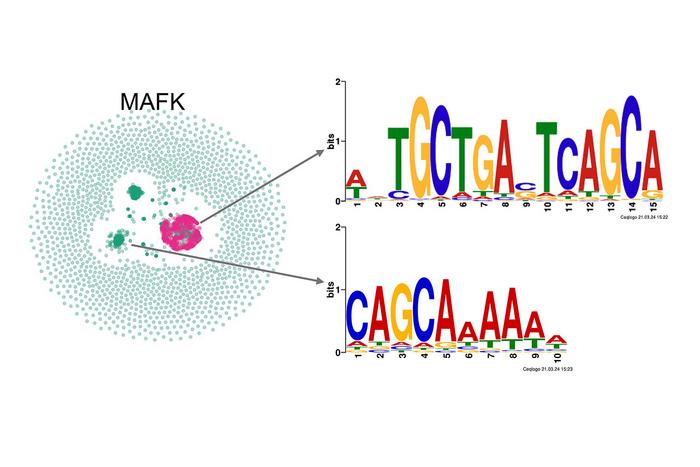

A groundbreaking computational method developed by Finnish scientists is poised to transform the way researchers analyze and visualize DNA sequence data. This innovative technique, known as k-mer manifold approximation and projection—or KMAP—is a powerful tool that translates complex genetic information into intuitive two-dimensional visual maps. By facilitating the exploration of DNA motifs and regulatory elements, KMAP offers a fresh lens through which molecular biologists can decode the intricate language of gene regulation.
The challenge of interpreting the vast amounts of data generated by sequencing technologies has long been a bottleneck in genomics research. DNA sequences are composed of short fragments called k-mers, which are strings of nucleotides of length k. Identifying biologically meaningful patterns within these short sequences is essential for understanding how genes are turned on or off in various contexts, including normal development and disease. KMAP addresses this challenge by projecting these k-mers onto a low-dimensional space that preserves meaningful relationships, allowing clusters representative of DNA motifs to emerge visually.
At the heart of KMAP is an advanced computational algorithm that leverages manifold learning principles. This approach captures the underlying geometry of the data by approximating the k-mer manifold—the shape that the high-dimensional k-mer data inhabits—and subsequently projecting it into two dimensions. Unlike traditional motif-finding tools that rely heavily on pre-defined models or heuristic searches, KMAP enables an unbiased and exploratory analysis. Each point in the resulting visualization corresponds to a single k-mer, with clusters delineating recurring sequence motifs observed in the genomic data.
One compelling application of KMAP involved the re-analysis of epigenomic data associated with Ewing sarcoma, a rare and aggressive pediatric cancer. The research team utilized KMAP to investigate the dynamic interactions of transcription factors within regulatory DNA regions of cancer cells. They discovered that upon degradation of the oncogenic transcription factor ETV6, other transcription factors such as BACH1, OTX2, and KCNH2/ERG1 became active predominantly at promoter and enhancer regions. This finding elucidates the complex transcriptional rewiring that occurs during tumorigenesis and underscores the importance of contextual motif activity.
Furthermore, KMAP uncovered a previously uncharacterized DNA motif defined by the sequence CCCAGGCTGGAGTGC. This novel motif was found to consistently co-localize with known factors BACH1 and OTX2 within enhancer regions, suggesting the presence of a collaborative regulatory element. The spatial proximity of these motifs hints at coordinated control mechanisms governing gene expression in cancer cells, opening new avenues for therapeutic targeting and biomarker discovery.
Beyond cancer genomics, KMAP shows immense potential in genome editing research. The team applied the method to analyze sequence repair outcomes following CRISPR-Cas9-mediated DNA cleavage at the AAVS1 locus in human cells. DNA repair is inherently variable, involving different pathways that result in distinct sequence alterations. By mapping thousands of DNA sequences obtained post-editing, KMAP visualized four major repair patterns, each linked to a specific cellular repair pathway. This insight empowers researchers to predict editing outcomes with greater accuracy, facilitating the design of more precise and efficient gene-editing interventions.
The intuitive visual nature of KMAP democratizes data interpretation for researchers who may not have extensive computational backgrounds. By converting high-dimensional sequence data into accessible graphics, the tool enables biologists to detect subtle regulatory motifs and contextual changes across diverse biological states. “KMAP offers a more intuitive way to investigate motifs in DNA sequence data,” explains Dr. Lu Cheng, lead author from the University of Eastern Finland. “By visualizing the distribution of short DNA sequences, we can better interpret regulatory patterns and understand how they change in different biological conditions.”
Professor Gonghong Wei of the University of Oulu highlights the versatility of KMAP. “This method is widely applicable, not only for identifying regulatory motifs from ChIP-seq datasets in cancer research but also for elucidating RNA-binding protein preferences and other sequence-centric molecular interactions. Its ability to reveal structure in complex sequence data provides a broadly useful computational framework across molecular biology.”
KMAP’s utility also extends to the study of transcription factor binding dynamics and epigenetic regulation. Since many biological processes depend on the interplay between multiple regulatory elements, this visualization method provides a comprehensive view of sequence motifs as interactive clusters, reflecting their spatial and functional relationships within the genome. Such detailed insight is invaluable for unraveling complex gene regulatory networks underlying health and disease.
The development of KMAP underscores the growing synergy between computational biology and experimental genomics. As sequencing technologies continue to generate unprecedented volumes of data, tools like KMAP are crucial for distilling actionable knowledge from genetic noise. Its capacity to integrate diverse sequencing data streams and deliver intuitive, interactive visualizations accelerates discovery and fosters deeper mechanistic understanding.
Importantly, KMAP is designed with accessibility and adaptability in mind. The software supports various input data types from sequencing experiments, making it an attractive resource for laboratories worldwide aiming to decipher regulatory codes in genomes. It also offers promising prospects for integration with other bioinformatics pipelines, thereby expanding its role in comprehensive genomic analyses.
In summary, KMAP represents a bold stride in computational genomics, enabling researchers to visually mine the manifold of k-mer sequences and extract biologically vital motifs with clarity and precision. This tool not only enhances motif discovery but also provides fresh perspectives on gene regulation dynamics across diverse biological processes, including cancer progression and genome editing. By bridging the gap between complex sequence data and meaningful biological interpretation, KMAP stands to become an indispensable asset in the molecular biology toolkit.
Subject of Research: Not applicable
Article Title: k-mer manifold approximation and projection for visualizing DNA sequences
News Publication Date: 10-Apr-2025
Web References:
DOI: 10.1101/gr.279458.124
Image Credits: Lu Cheng
Keywords:
Gene regulation
DNA sequences
Computational biology
Tags: advanced data visualization methodscancer genomics researchcomputational biology toolsDNA regulation mechanismsDNA sequence interpretationgene regulation analysisgenome editing techniquesinterpreting sequencing datak-mer manifold approximationmanifold learning applicationsmolecular biology innovationsvisualizing genetic data



{kind=link}