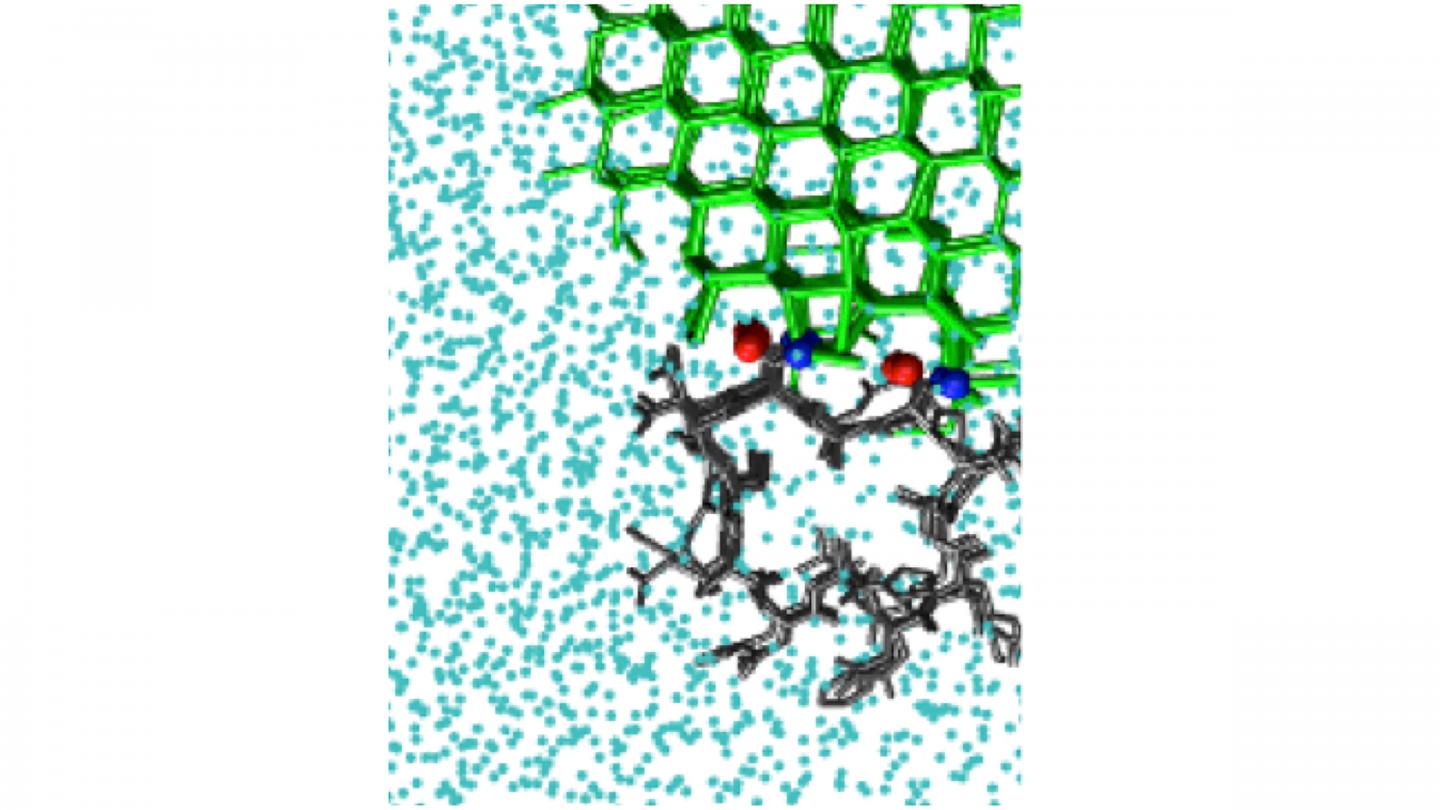
Credit: Pavithra M. Naullage
WASHINGTON, November 3, 2020 — Certain molecules bind tightly to the surface of ice, creating a curved interface that can halt further ice growth. Some insects, plants, and sea-dwelling creatures contain protein molecules of this type that act as natural antifreeze agents, allowing the organisms to withstand freezing temperatures.
In The Journal of Chemical Physics, by AIP Publishing, scientists report a computational method to model ice binding using a biasing technique to drive the formation of ice in the simulation.
Antifreeze proteins act by binding to an existing interface between ice and liquid water. The resulting curved surface stops the growth of ice. There are also ice-nucleating molecules that catalyze the formation of ice from supercooled liquid water.
Both phenomena require an understanding of the way that molecules bind to ice. Understanding ice binding is important for applications as diverse as cryopreservation of organs and climate modeling, but no computational methods to efficiently model this phenomenon have existed to date.
“The central advantage of the ice-biasing simulation approach is that it simultaneously identifies the ice-binding surface, the face of ice it binds to, and the mode of binding,” said author Valeria Molinero.
The investigators created two types of models. One type is an all-atom model that contains all the atoms in the liquid and ice phases of water as well as in the antifreeze-type molecule. The other type of model studied is called a coarse-grained model, which saves computational resources by blending atoms together into simpler structures.
The study looked at a number of molecules that bind ice, including polyvinyl alcohol, a synthetic ice-recrystallization inhibitor, as well as natural antifreeze proteins, such as one from the beetle Tenebrio molitor. Proteins present a simulation challenge, since they have very small surfaces that bind ice. This limits the size of the ice crystal they can bind.
Some systems possess more than one location where ice can bind. This is the case for the natural antifreeze protein in the sea-ice diatom Frailariopsis cylindrus. To determine whether a protein like this has more than one ice-binding surface, IBS, the investigators developed a method they dubbed “cap and repeat.”
“In this strategy, we first performed a biased simulation to detect an IBS. Then, we cap that IBS to prevent ice formation on it and perform a second biasing simulation to find out whether ice forms in other sites,” said Molinero.
The methods developed in this study show great promise for a number of applications, including finding molecules to protect frozen tissues during storage, furthering the understanding of natural antifreeze proteins, and in climate models, where ice nucleation in the atmosphere plays a key role.
###
The article, “Computationally efficient approach for the identification of ice-binding surfaces and how they bind ice,” is authored by Pavithra M. Naullage, Atanu K. Metya, and Valeria Molinero. The article will appear in The Journal of Chemical Physics on Nov. 3, 2020 (DOI: 10.1063/5.0021631). After that date, it can be accessed at https:/
ABOUT THE JOURNAL
The Journal of Chemical Physics is an international journal that publishes cutting edge research in all areas of modern physical chemistry and chemical physics. See https:/
Media Contact
Larry Frum
[email protected]
Related Journal Article
http://dx.

{kind=link}