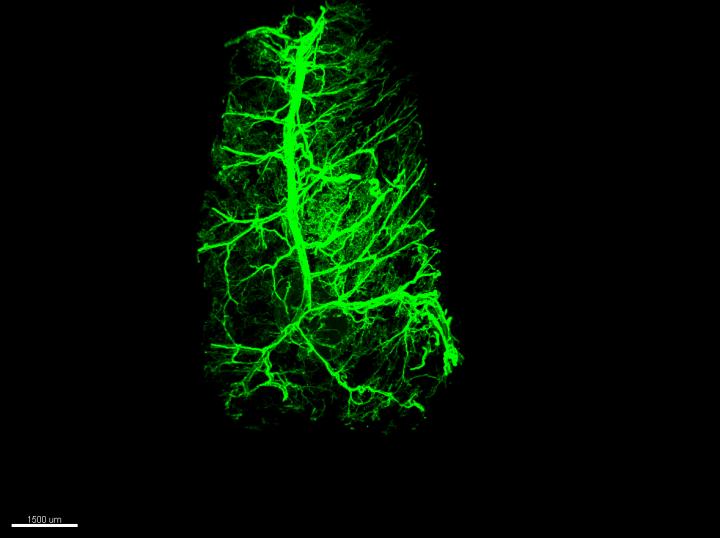
Credit: Laboratory of Molecular Genetics at The Rockefeller University
There’s no doubt that you can lose fat by eating less or moving more–yet after decades of research, the biology underlying this equation remains mysterious. What really ignites the breakdown of stored fat molecules are nerves embedded in the fat tissue, and a new study now reveals that these fat-burning neurons have previously unrecognized powers. If they receive the right signal, they have an astonishing capacity to grow.
That signal is the hormone leptin, which is released by the fat cells themselves. In experiments with mice described on July 22 in the journal Nature, the researchers found that the normally bushy network of neural fibers within fat tissue shrinks in the absence of leptin and grows back when the hormone is given as a drug. These changes were shown to influence the animals’ ability to burn the energy stored in fat.
“While the architecture of the nervous system can change significantly as a young animal develops, we did not expect to find this profound level of neural plasticity in an adult,” says . Jeffrey M. Friedman, a molecular geneticist at The Rockefeller University.
If confirmed in humans, the findings could advance research on obesity and related diseases, and potentially open the door to developing new treatments that target neurons in fat.
Homing in on neurons in fat
The team began by looking at what happens to mice who do not produce leptin on their own, and how they respond when treated with it.
Discovered in Friedman’s lab in 1994, the hormone relays signals between fat deposits and the brain, allowing the nervous system to curb appetite and boost energy expenditure to regulate body weight. When mice are genetically engineered to stop producing leptin, they grow three times heavier than normal mice. They eat more, move less, and cannot survive in what should be tolerable cold because their body can’t properly utilize fat to generate heat.
Give these mice a dose of leptin, however, and they quickly begin to eat less and move more. But when the researchers treated them longer, for two weeks, more profound changes occurred: the animals started to break down white fat, which stores unused calories, at normal levels, and regained the ability to use another form of fat tissue, brown fat, to generate heat.
It was this slower change that interested the research team, including the first authors of the Nature paper, Putianqi Wang, a graduate student in the lab, and Ken H. Loh, a postdoctoral fellow. They suspected that changes in neurons outside the brain–those that extend into fat–might explain why this part of the response to leptin took some time.
To the brain and back
Using an imaging technique developed by the lab of Rockefeller’s Paul Cohen to visualize nerves inside fat, the researchers traced leptin’s effects on the fat-embedded neurons up to the brain’s hypothalamus region. From here, they found, leptin’s growth-promoting message travels via the spinal cord back to the neurons in fat. “This work provides the first example of how leptin can regulate the presence of neurons in fat, both white and brown,” adds Cohen.
Through this pathway, fat appears to be telling the brain how much innervation it needs to function properly. “Fat is indirectly controlling its own innervation and thus function,” Friedman says. “It is an exquisite feedback loop.”
Future research will analyze the role of this pathway in human obesity and possibly provide a novel approach for therapy. Most obese people produce high levels of leptin, and show a diminished response to hormone injections, suggesting that their brain is resistant to the hormone. Thus, bypassing leptin resistance could have a therapeutic benefit for these patients. “In the new study we see that similar to animals lacking leptin, obese, leptin-resistant animals also show reduced fat innervation,” Friedman says. “So we speculate that directly activating the nerves that innervate fat and restoring a normal ability to use stored fat could provide a possible new avenue for treating obesity.
###
Media Contact
Katherine Fenz
[email protected]
Related Journal Article
http://dx.



{kind=link}