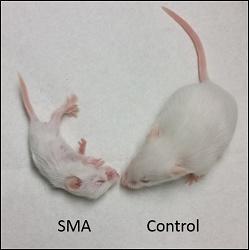
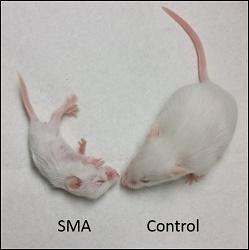
Credit: Sumner lab
Johns Hopkins researchers along with academic and drug industry investigators say they have identified a new biological target for treating spinal muscular atrophy. They report they have evidence that an experimental medicine aimed at this target works as a "booster" in conjunction with a drug called Nusinersen that was recently FDA-approved to improve symptoms of the disorder in mice.
In a report on the work, published in Neuron on Dec. 23, the researchers say the combination therapy improved survival time, body weight and motor movements in mice with spinal muscular atrophy, a relatively rare inherited disorder characterized by the loss of motor neurons, or nerve cells that control movement.
Nusinersen, approved by the U.S. Food and Drug Administration in December 2016, became the first drug approved to treat spinal muscular atrophy, with several other experimental drugs still in clinical trials. The Johns Hopkins researchers and their colleagues say the "boosted" nusinersen therapy, if confirmed by further studies, has the potential to significantly improve the lives of people with spinal muscular atrophy, some with forms of the disease with only weeks or months to live, and others who are unable to sit, stand or walk.
The researchers caution that the booster therapy used in their new study will not be available on the market or even for use in human trials anytime soon; it must await years of animal testing for safety and effectiveness first. But the results of their study, they add, offer at least one path to extending the usefulness of nusinersen.
"The new FDA-approved drug nusinersen may have a 'ceiling effect,' in that it may not completely treat the disorder," says Charlotte Sumner, M.D., associate professor of neurology at the Johns Hopkins University School of Medicine. "Our approach was to work with those that developed nusinersen to identify a way to boost their drug's effects on the sickest of the sick."
Spinal muscular atrophy, one of the leading genetic killers of infants, arises from not having enough survival motor neuron (SMN) protein in neurons. The disorder is due to a genetic mutation in the SMN1 gene that is the blueprint for SMN, a motor neuron protein important for development. One in 40 people carry the defective gene for spinal muscular atrophy, and as many as one in 6,000 babies in the world across all ethnic groups is born with the disorder.
When looking into mechanisms that might affect the levels of SMN protein in neurons, the researchers scanned a genomic database called the UCSC Genome Browser and identified two genetic sequences that matched the opposite DNA strand of the SMN gene. Using programs that analyzed the DNA sequences, they determined that they made up one long RNA. This type of RNA was different, though, in that it didn't make up or code for the instructions of a protein like a traditional gene but instead was "noncoding."
The researchers detected this SMN long noncoding RNA, or lnc-RNA (pronounced "link RNA") for short, in human embryonic kidney cells, brain cell samples and neurons derived from the stem cells of healthy people and those with spinal muscular atrophy type I and II. In healthy people and mice, the researchers observed the SMN protein at high levels before birth and then decreasing after birth. The researchers noticed the opposite pattern with the SMN lnc-RNA; it was at low levels before birth but then increased after birth in human spinal cord samples. They confirmed these same opposite patterns of SMA protein and SMN lnc-RNA in embryonic mice too.
Then, researchers then sought a way to increase SMA gene product levels, the messenger RNAs (mRNAs), and thus protein levels by silencing or removing the SMN lnc-RNA. To do that, they created eight different bits of DNA that matched the opposite strand, or antisense version of the SMN lnc-RNA, which would bind to the lnc-RNA and cause it to break down.
After incubating HeLa cells and neurons from embryonic mice with different amounts of these DNA opposite strand bits, they noted that the more DNA bits they added, the more mRNA levels increased that code for SMA protein. Two of these DNA bits, named ASO-A and ASO-B, were the most effective. These and other experiments suggested that the SMN lnc-RNA seems to turn down levels of the SMN mRNA, thus preventing more SMN protein from being made.
Nusinersen and similar compounds being developed for spinal muscular atrophy focus on increasing or making more of the SMN gene product that gets converted into usable protein. To see if shutting down the SMN lnc-RNA could increase SMN levels and slow or stop spinal muscular atrophy, the researchers injected 400 milligrams per kilogram of ASO-A DNA bits into newborn mice with severe spinal muscular atrophy at day one and day three after birth. At day 10, they researchers detected a 70 percent reduction of SMN lnc-RNA in the brains of treated mice, but survival, body weight and the ability to get on their feet wasn't improved compared to mice injected with saline.
The first and newest FDA-approved treatment of nusinersen doesn't completely cure the disease, so Sumner's research team wanted to see if using ASO-A in conjunction with nusinersen would further improve muscle form and function in mice with spinal muscular atrophy. Nusinersen is also made of small pieces of DNA that changes how the SMN mRNA is processed so that more is made into protein.
Mice with spinal muscular atrophy were injected with ASO-A alone, a low 50 milligram per kilogram dose of nusinersen alone, both ASO-A and nusinersen, or saline at day one after birth. Fifteen mice received each treatment. The combination therapy increased the average survival to 37 days, with four of the mice living more than 120 days.
Saline and ASO-A mice lived an average of 18 days, and nusinersen only-treated mice lived an average of 25 days. Mice given both drugs also reached an average body weight of 14 grams after 50 days. Mice with only nusinersen didn't gain much weight past 6 grams. Healthy mice without the disease weigh about 20 grams after 50 days.
The researchers also tested the mice on the number of times they reared up on their hind legs within a minute of being put in a clear cylinder, a standard experimental measure of motor ability. Healthy mice will rear about six times, mice with spinal muscular atrophy treated with both therapies reared about four times and mice with either drug alone only reared about two times.
"We don't know yet if the SMN lnc-RNA actually plays a role in the disease, but it is a convenient target that we've used to our advantage to treat spinal muscular atrophy in mice," says Sumner. "We still want to determine how the lnc-RNA does its job and when it does it job, and that will give us a better handle on how to target it more effectively."
Sumner says that lnc-RNAs are still a rapidly emerging field, and we are just beginning to learn what they do and their role in disease.
Biogen, which makes nusinersen, has set the price per injection of nusinersen at $125,000, or about $750,000 for the first year of treatment. The treatment won't be mass-produced because spinal muscular atrophy is a rare disease.
Additionally, because the drug doesn't cross the blood-brain barrier, it needs to be administered through a spinal tap, which requires specialized facilities. The most common side effects of the drug include respiratory infections and constipation, with possible chances of reduced platelet levels and kidney damage. Long-term or lasting side effects, particularly if the treatments affect other tissues outside the nervous system, have yet to be determined.
###
Additional authors on the paper include Constantin d'Ydewalle, Daniel Ramos, Noah Pyles, Mariusz Gorz, Celeste Pilato and Lingling Kong of The Johns Hopkins University; Shi-Yan Ng and Lee Rubin of Harvard University; Karen Ling, Frank Rigo and C. Frank Bennett of Ionis Pharmaceuticals; and Amanda Ward of the Massachusetts Institute of Technology.
The study was funded by the SMA Research Team, the Muscular Dystrophy Association, the American Association of Neuromuscular and Electrodiagnostic Medicine, FightSMA Inc., the Gwendolyn Strong Foundation, and grants from the National Institute on Neurological Disorders and Stroke (R01 NS096770) and the National Center for Advancing Translational Sciences (UL1 TR 001079).
Media Contact
Vanessa McMains
[email protected]
410-502-9410
@HopkinsMedicine
http://www.hopkinsmedicine.org
############
Story Source: Materials provided by Scienmag


{kind=link}