A new statistical framework promises sharper answers to a long-standing question in conservation and evolutionary genetics: who is a hybrid, and who is a backcross in the wild? In a study published this week, researchers present a Bayesian hybrid inference method that leverages sampled genomes from two populations across two generations. The goal is to assign individuals probabilistically to classes such as hybrids or backcrosses while respecting the complexities of real genomes.
The approach builds on an earlier method by Chakraborty and Rannala (2023), but it introduces a crucial upgrade: it explicitly accounts for uncertainty in population haplotype frequencies. That matters because haplotype frequencies inferred from finite samples are never known exactly. Treating them as fixed quantities can inflate confidence, especially when datasets are small—precisely the scenario faced by many non-model organisms.
Technically, the new framework improves inference by correctly marginalizing over haplotypes while still modeling genetic linkage and recombination along chromosomes. Rather than breaking the genome into independent pieces, it retains the dependence created by shared ancestry and recombination events. This allows the method to extract more informative signals than analyses that ignore linkage structure.
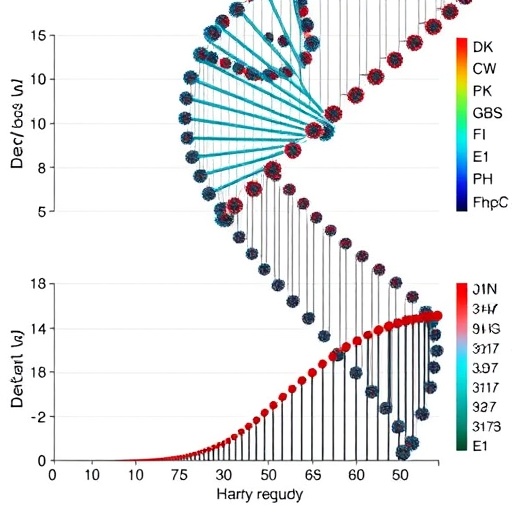
Simulations show that when the number of individuals sampled to estimate population haplotypes is large, posterior probabilities from the new method nearly match those produced by the earlier 2023 approach. But when sample sizes shrink, the new method’s posterior probabilities decline, reflecting a more conservative—and more realistic—handling of uncertainty.
Performance benchmarks using ROC (Receiver Operating Characteristic) curves indicate that predictive discrimination remains essentially equivalent to the earlier method. In other words, the upgrade does not appear to sacrifice the ability to separate true hybrids from non-hybrids; it mainly adjusts the degree of certainty.
To test generality beyond simulations, the team applied the method to three recently published datasets spanning three very different taxa. The results were evaluated in kiwifruit (Actinidia), the plateau fence lizard (Sceloporus tristichus), and the puma (Puma concolor), illustrating the method’s versatility.
By providing a principled way to infer hybridization and backcrossing while reflecting uncertainty in population-level genetic summaries, the framework could help researchers interpret contact zones, manage breeding plans, and untangle introgression histories with fewer overconfident conclusions.
Overall, the work signals a shift toward hybrid-detection tools that are both statistically rigorous and computationally aligned with the realities of genomic data—where the genome is linked, recombination matters, and uncertainty cannot be ignored.
Subject of Research: Hybrid and backcross inference using genome sequences across two generations
Article Title: Improved Bayesian inference of hybrids using genome sequences
Article References: Chakraborty, S., Rannala, B. Improved Bayesian inference of hybrids using genome sequences. Heredity (2026). https://doi.org/10.1038/s41437-026-00861-6
DOI: 10.1038/s41437-026-00861-6
Keywords: Bayesian inference, hybridization, backcrossing, haplotypes, linkage and recombination, ROC performance, genome sequences
Tags: Bayesian hybrid inferenceconservation geneticsevolutionary geneticsfinite sample effects on haplotype frequency estimationgenome sequence data analysisGenomic hybrid detectionimproved genetic assignment methodsincorporating linkage disequilibrium in inferencelinkage and recombination modelingpopulation haplotype frequency uncertaintyprobabilistic classification of hybrids and backcrossesstatistical framework for hybrid identification


{kind=link}