Credit: Chi Chen/Materials Virtual Lab
Nanoengineers at the University of California San Diego have developed new deep learning models that can accurately predict the properties of molecules and crystals. By enabling almost instantaneous property predictions, these deep learning models provide researchers the means to rapidly scan the nearly-infinite universe of compounds to discover potentially transformative materials for various technological applications, such as high-energy-density Li-ion batteries, warm-white LEDs, and better photovoltaics.
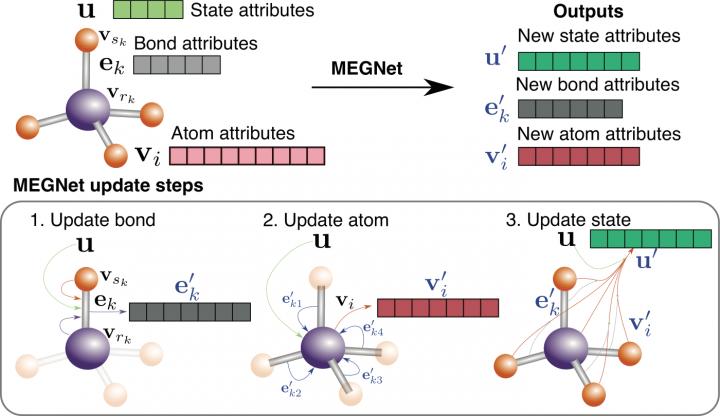
To construct their models, a team led by nanoengineering professor Shyue Ping Ong at the UC San Diego Jacobs School of Engineering used a new deep learning framework called graph networks, developed by Google DeepMind, the brains behind AlphaGo and AlphaZero. Graph networks have the potential to expand the capabilities of existing AI technology to perform complicated learning and reasoning tasks with limited experience and knowledge–something that humans are good at.
For materials scientists like Ong, graph networks offer a natural way to represent bonding relationships between atoms in a molecule or crystal and enable computers to learn how these relationships relate to their chemical and physical properties.
The new graph network-based models, which Ong’s team dubbed MatErials Graph Network (MEGNet) models, outperformed the state of the art in predicting 11 out of 13 properties for the 133,000 molecules in the QM9 data set. The team also trained the MEGNet models on about 60,000 crystals in the Materials Project. The models outperformed prior machine learning models in predicting the formation energies, band gaps and elastic moduli of crystals.
The team also demonstrated two approaches to overcome data limitations in materials science and chemistry. First, the team showed that graph networks can be used to unify multiple free energy models, resulting in a multi-fold increase in training data. Second, they showed that their MEGNet models can effectively learn relationships between elements in the periodic table. This machine-learned information from a property model trained on a large data set can then be transferred to improve the training and accuracy of property models with smaller amounts of data–this concept is known in machine learning as transfer learning.
###
The study is published in the journal Chemistry of Materials. The MEGNet models and software are available open-source via Github (https:/
Paper title: “Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals.” Co-authors include Chi Chen, Weike Ye, Yunxing Zuo and Chen Zheng, UC San Diego.
This work is supported by the Samsung Advanced Institute of Technology (SAIT) Global Research Outreach (GRO) Program, utilizing data and software resources developed by the DOE-funded Materials Project (DE-AC02-05-CH11231). Computational resources were provided by the Triton Shared Computing Cluster (TSCC) at UC San Diego; the Comet supercomputer, a National Science Foundation-funded resource at the San Diego Supercompuer Center (SDSC) at UC San Diego; the National Energy Research Scientific Computer Centre (NERSC); and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation (Grant No. ACI-1053575).
Media Contact
Liezel Labios
[email protected]
Related Journal Article
http://dx.



{kind=link}