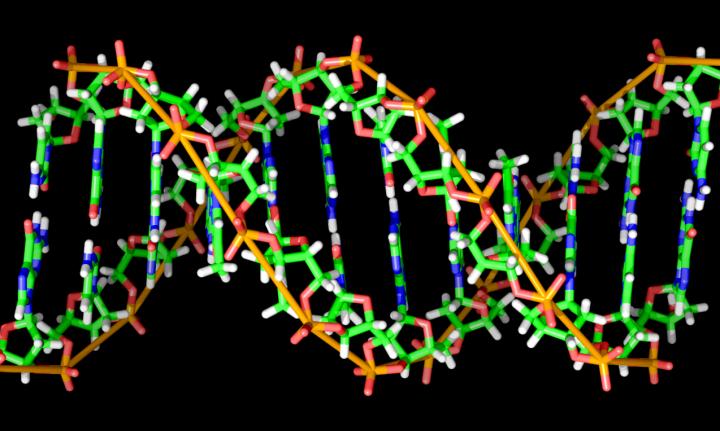
Credit: Richard Wheeler (Zephyris). Used under the Creative Commons Attribution-ShareAlike 3.0 license.
In the same way that barcodes on your groceries help stores know what's in your cart, DNA barcodes help biologists attach genetic labels to biological molecules to do their own tracking during research, including of how a cancerous tumor evolves, how organs develop or which drug candidates actually work. Unfortunately with current methods, many DNA barcodes have a reliability problem much worse than your corner grocer's. They contain errors about 10 percent of the time, making interpreting data tricky and limiting the kinds of experiments that can be reliably done.
Now researchers at The University of Texas at Austin have developed a new method for correcting the errors that creep into DNA barcodes, yielding far more accurate results and paving the way for more ambitious medical research in the future.
The team — led by postdoctoral researcher John Hawkins, professor Bill Press and assistant professor Ilya Finkelstein — demonstrated that their new method lowers the error rate in barcodes from 10 percent to 0.5 percent, while working extremely rapidly. They describe their method, called FREE (filled/truncated right end edit) barcodes, today in the journal Proceedings of the National Academy of Sciences.
The researchers have applied for a patent and are making the method freely available for academic and noncommercial use.
With DNA barcodes, scientists can study how a cancerous tumor evolves, not just as a whole, but as a large collection of individual cells that evolve differently to reveal which cells are vulnerable to therapeutics and which aren't. Scientists interested in growing replacement organs for injured or sick people can use DNA barcodes to better understand how organs naturally develop. And researchers looking to screen millions of potential drugs to find one that binds to a certain molecule, and thus has the potential to treat a disease, can use DNA barcodes to find the proverbial needle in a haystack.
"DNA barcodes are a part of a great deal of cutting-edge research in medicine and drug development, and to be able to improve the accuracy and efficiency of so many of these is very exciting," said Hawkins. "And maybe even more exciting is that now with these better barcodes, this allows us to have larger, more ambitious experiments that weren't possible before."
A DNA barcode contains a short string of letters that equates to a unique code, using the four letters found in DNA: A, C, G and T. These barcodes are stuck onto molecules, such as cellular proteins or drug candidates, as a way of keeping track of where they all go, sometimes by the millions, and how they interact with other molecules. About one-tenth of the time, however, errors occur — such as one letter being replaced by the wrong letter, an extra letter being inserted, or a letter being deleted — potentially skewing the results of critical biomedical research.
One of the keys to this new error-correction method is to select just the right barcodes from the beginning. This method involves choosing a string of letters for each barcode such that even if a small error creeps in — say, a G is substituted for a C — it will still be more like the intended barcode than any other. The method requires throwing out many possible strings of letters, but the researchers minimized this loss by borrowing an approach from computer science called sphere packing.
"My contribution has been designing a way to find those barcodes such that even if there is an error in it, you know which original barcode it came from," Hawkins said.
Alternative error-correcting methods for DNA barcodes, such as what are known as Levenshtein codes, require throwing away up to 100 times as many barcodes as with the FREE method, and they are up to 1,000 times slower to decode the results. As a result, whereas existing technology made projects with hundreds of millions of barcodes nearly impossible, the new technology allows for rapid, accurate results.
###
Hawkins is a postdoctoral researcher in UT Austin's Department of Molecular Biosciences (MBS) and Institute for Computational Engineering and Sciences (ICES). Press is a professor in the Department of Integrative Biology, Department of Computer Science and ICES. Finkelstein is an assistant professor in MBS and the Center for Systems and Synthetic Biology.
This research was supported by a College of Natural Sciences Catalyst Award, as well as grants from the Welch Foundation and the National Institutes of Health.
MORE INFO
Computer scientists call problems such as these sphere-packing problems. A real-world example is finding a way to pack the most oranges into a crate.
Here's how sphere packing works for DNA barcodes: For each string of letters of a certain length that you could possibly make, you first make a list of all the possible barcodes you could make by introducing one or two changes. If you imagine each barcode as a point in three-dimensional space, these other nearly identical barcodes form a cloud around that point. That cloud of barcodes can be enclosed in a shape called a decode sphere. Then, just like packing oranges into a crate, you can use an algorithm designed to pack the most spheres into a given space. Solving that problem is the same as maximizing the number of error-correcting barcodes you can pluck out of the universe of all possible barcodes of a given length.
"For each barcode, I want to reserve all the words around that barcode that you can get to with a single error," John Hawkins said. "So if I pick the word AAA, then I also need to include in my sphere AAC. That's one change and so it's within a one-edit sphere. With barcodes, if you pack all the spheres into a space and none of them overlap, what that means is whenever you see a sequence with only one error, it's only in one of those spheres, so you know which sphere it is in. Therefore, you know which barcode was intended."
Media Contact
Marc Airhart
[email protected]
512-232-1066
@UTAustin
http://www.utexas.edu

{kind=link}