University of Washington’s Baker Lab uses de novo computational design on TACC systems to create new therapeutics
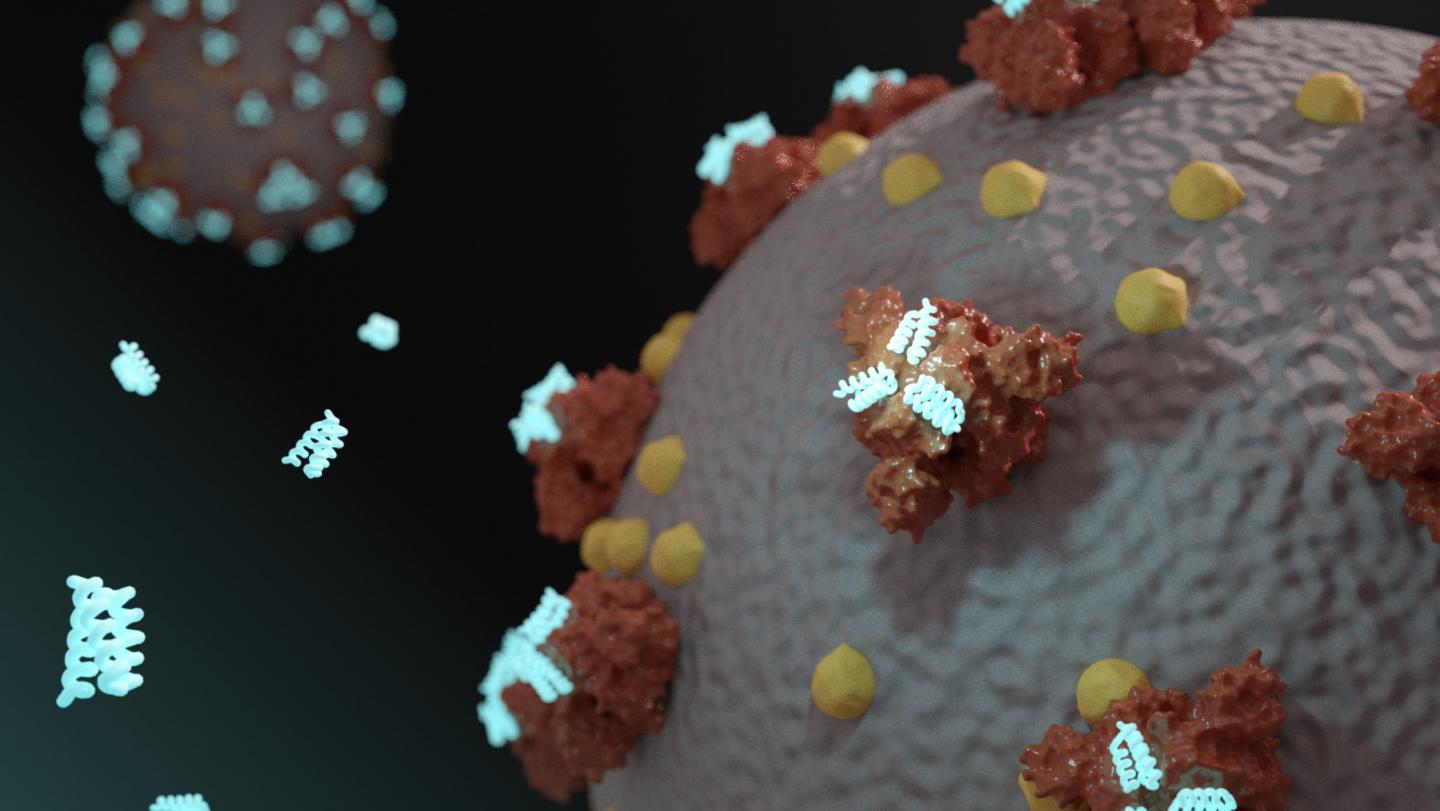
Credit: Ian C Haydon / Institute for Protein Design
Nearly every drug that is, or has ever been, used was derived from nature — harnessing compounds created by organisms over eons to fight diseases. But decades ago, biochemists postulated that it might be possible to design a new drug from scratch by linking up amino acids in precise ways.
The tricky part, as it turned out, was predicting in advance how the amino acids in a protein would fold. The folded form determines the three-dimensional shape of the protein, as well as its electrostatic potential, and hydrophobicity (the degree to which a molecule is repelled from a mass of water) — factors that are critical when it comes to designing an effective drug.
David Baker, professor of Biochemistry at the University of Washington and head of the Institute for Protein Design there, pioneered methods for using computers to predict how proteins fold. Based on that knowledge, he and his team have designed new, never-before-seen proteins for use as drugs, sensors, or even biological logic gates.
This approach is known as de novo protein design. There are currently only a few drugs in trial that have used this approach, but it holds incredible potential — and at no time has such an approach been more critical than now.
The platform that the Institute for Protein Design developed allows for the rapid design of protein binders to target proteins of interest. Computer (in silico) simulations generate a library of candidate protein sequences that are then tested at their in-house testing facility. Promising candidates are evolved both in silico and in the wet lab until a final binding protein is created.
Starting in January, researchers in the Baker Lab began have been using their methodology to design a drug or vaccine to treat COVID-19. Their studies involve calculating the three-dimensional shape of millions of possible proteins, and then computationally testing how such proteins would fit into, and dock with, parts of the SARS-CoV-2 virus.
To assist in this effort, they are using the Stampede2 supercomputer at the Texas Advanced Computing Center (TACC) — one of the fastest in the world — as well as the network of volunteer computers known as Rosetta@Home. (Rosetta is the name of the software developed in the Baker Lab to predict protein folding and to design new proteins.)
“In just a two-month span, our team has been able to computationally design millions of protein therapeutics that target the seven major proteins related to COVID-19,” Baker reported in March.
To date, 733,000 proteins have been ordered, 323,000 of these protein therapeutics have been tested in the laboratory, and more than 2,000 have shown binding signals to their respective targets.
From Scaffolds to Protein Structures
The team began by testing its collection of 20,000 scaffold proteins that form the starting point for future drugs or vaccines. Each can be docked in over 1,000 orientations; and each dock is subsampled 1,000 times with slight perturbations – leading to 20 billion potential interactions to compute.
“In the scaffold phase, we’re looking for signs these are going to be atomically accurate,” said Brian Coventry, a PhD student in the group working on the project. “If we’re off by 0.1 nanometers, there’s no way it will work. These things have to be perfect.”
The top 1 million of these docks then move forward to sequence design where each position on the scaffold backbone must be assigned an amino acid. With 20 amino acid choices at each position, and a variety of conformations for each, the computer must solve the combinatorial explosion to assign the best combination of amino acids to each scaffold.
From the 1 million designed proteins, they determine the most promising subset – roughly 100,000 proteins. The team sends a text file containing DNA sequences for these candidates to Agilent, a company that can create synthetic DNA molecules on demand. Agilent returns test tubes with physical DNA, which is then inserted into yeast genomes in such a way that the various synthetic proteins are made and displayed on tethers from the cell membrane of yeast, allowing them to be tested against the virus.
Based on the initial computational and experimental results, the team then engages in site saturation mutagenesis, where each individual amino acid on the chain is mutated at every location and re-tested to see how it behaves.
“We get data back and look at what made a given protein better or worse. And we ask the question: ‘Does this protein look like it’s working for the right reasons?'” Coventry said.
Based on the results and insights from the mutagenesis, they go one step further and develop a combo library that includes degenerate codons, where alternate nucleotides replace the typical ones in a given amino acid.
The best combination of mutations and replacements undergo further experimental testing including bacterial expression and thermodynamic analysis. Using this method, they derived 50 highly promising leads for the spike protein binder from an initial screening of 100,000 proteins.
“The spike protein binder is the most likely to result in a drug because of its mechanism of action,” Coventry said.
But the ability to create designer proteins is not the lab’s only innovation, nor is a single binder their final goal. They are also pioneering a new approach to drugs called mini-protein binders that combine the specificity of antibodies with the high stability and manufacturability of small molecule drugs.
Mini-protein binders have been shown to have much greater stability at elevated temperatures and better neutralization than comparable antibodies and natural protein derivatives. They are also approximately 1/30th of the molecular weight of typical proteins, and can be synthesized chemically, which enables the introduction of a wide variety of functionalities. Probably as a result of their small size and very high stability, they elicit little immune response.
“We aim to connect four to six of the most potent neutralizers in a single chain by flexible linkers to achieve highly avid binding with little potential for escape,” Baker said in a presentation to the Defense Advanced Research Projects Agency (DARPA), one of the funders of the research.
“We try to get many binders and connect them with linkers,” Coventry explained further. “The idea is that you get an avidity effect” — the accumulated strength of multiple affinities. “At least one of those proteins will be binding at any given time and the virus particle won’t be able to escape the chain. Since the binders block the viral binding epitope, the virus will not be able to enter our cells.”
Building on Collaborations
TACC is currently supporting more than 40 COVID-19 research projects. The one from the Baker Lab has been among the largest users of compute time on Stampede2 since it began in March.
“TACC has a lot of computing power and that has been really helpful for us,” Coventry said. “Everything we do is purely parallel. We’re able to rapidly test 20 million different designs and the calculations don’t need to talk to each other.” This type of approach, known as high-throughput screening, is a good fit for Stampede2’s architecture.
Baker and his team were able to ramp up quickly on TACC resources in part because of their involvement in an ongoing DARPA-funded program known as the Synergistic Discovery and Design (SD2), a multi-institution collaboration whose goal is to develop data-driven methods to accelerate scientific discovery.
Since 2017, the SD2 program has been developing pipelines to “design-test-learn” faster, using a combination of high-performance computing, advanced data management practices, automated laboratory testing, and machine learning. The collaboration between the Baker Lab and TACC is emblematic of that methodology and is helping to accelerate their research from idea to reality.
According to Dr. Matthew Vaughn, Director of Life Sciences Computing at TACC, the protein design project appears poised to yield powerful new therapeutic molecules for the fight against COVID-19 due in part to the remarkable synergy between computational simulation and experimentation.
“The rapid pace at which the Baker lab has been able to onboard and become productive on a leadership-class resource like Stampede 2 reinforces just how critical our national investments in advanced computing capability and methodology have been and will continue to be in the future,” Vaughn said.
The team’s next milestone will be to develop multiple inhibitors that can reduce the response by half and that can be linked those together into a big molecule, or construct, that is well behaved.
Further testing to establish whether the mini-binder provokes an immune response would follow, and then the construct would be tested for efficacy in a petri dish, then in animals and humans.
“Our goal for the next pandemic will be to have computational methods in place that, coupled with high performance computing centers like TACC, will be able to generate high affinity inhibitors within weeks of determination of the pathogen genome sequence,” Baker said. “To get to this stage will require continued research and development, and centers like TACC will play a critical role in this effort as they do in scientific research generally.”
Media Contact
Aaron Dubrow
[email protected]
Original Source
https:/



{kind=link}