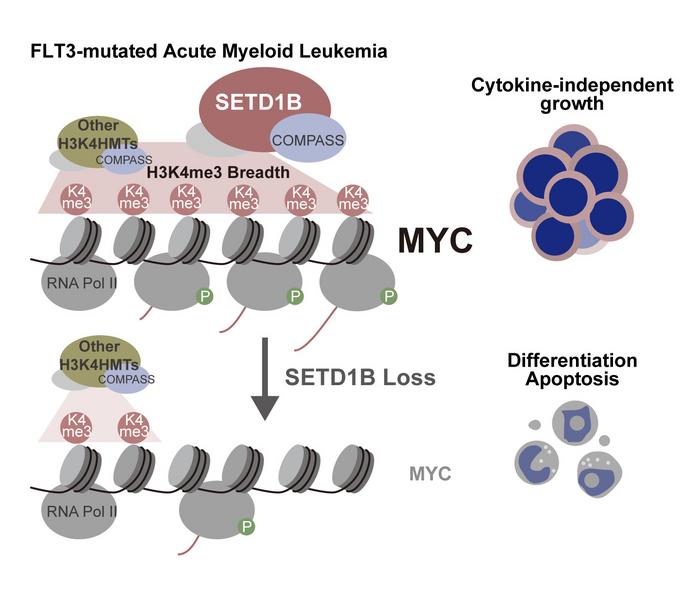
Acute myeloid leukemia (AML) remains one of the most formidable hematological malignancies, characterized by the rapid proliferation of malignant myeloid cells within the bone marrow and peripheral blood. Despite advances in chemotherapy and targeted therapeutics, many subtypes of AML resist treatment, leading to frequent relapses and poor patient outcomes. A groundbreaking study conducted by researchers at Chiba University in Japan has now illuminated an epigenetic mechanism that fuels the aggressive growth of AML harboring the FLT3 internal tandem duplication (FLT3-ITD) mutation, offering a new avenue for therapeutic intervention.
This recent research uncovers the pivotal role of the epigenetic enzyme SETD1B in driving the malignant expansion of leukemia cells, particularly those with the FLT3-ITD mutation. FLT3-ITD mutations are notorious for conferring a higher risk of relapse and decreased overall survival, yet the molecular basis underpinning their aggressiveness remained ambiguous. The team’s findings reveal that SETD1B exerts its oncogenic influence by broadening the landscape of tri-methylated histone H3 lysine 4 (H3K4me3) marks—an epigenetic modification linked to active transcription—and by sustaining elevated levels of the proto-oncogene MYC, which is a key regulator of cell proliferation and metabolism.
Histone modifications are subtle yet powerful epigenetic signals that alter chromatin structure and gene expression without changing the underlying DNA sequence. In mixed lineage leukemia rearranged (MLL-r) AML, high levels of H3K4me3 have been observed, but prior to this study, the connection between FLT3-ITD mutations and epigenetic landscapes had not been clearly defined. Using state-of-the-art CRISPR screening and chromatin immunoprecipitation coupled with RNA sequencing, the researchers meticulously mapped how SETD1B controls the breadth of H3K4me3 marks and consequently regulates MYC transcription.
.adsslot_ZFGaJb5NT6{ width:728px !important; height:90px !important; }
@media (max-width:1199px) { .adsslot_ZFGaJb5NT6{ width:468px !important; height:60px !important; } }
@media (max-width:767px) { .adsslot_ZFGaJb5NT6{ width:320px !important; height:50px !important; } }
ADVERTISEMENT
The experimental design included engineering leukemia cells to conditionally delete the catalytic domain of SETD1B, effectively disabling the enzyme’s ability to deposit methyl groups onto histone H3 at lysine 4. Loss of this catalytic activity resulted in a marked deceleration of leukemic cell growth, especially in cells carrying the FLT3-ITD or Nras^G12D mutations, two drivers of AML pathogenesis. Intriguingly, this epigenetic disruption also led to a downregulation of genes within the MYC signaling pathway, highlighting a direct mechanistic link between SETD1B enzymatic function and oncogenic transcriptional programs.
Further illuminating the biological significance of their findings, the team demonstrated that reintroducing MYC into SETD1B-deficient leukemia cells partially restored their proliferative capacity. This partial rescue underscores that SETD1B’s oncogenic influence extends beyond mere regulation of MYC expression and suggests that other downstream pathways or epigenetic marks governed by SETD1B may contribute to leukemia progression. As Associate Professor Takayuki Hoshii, lead author of the study, explains, the maintenance of H3K4me3 breadth ensures transcriptional consistency and the robustness of MYC expression, both essential for sustaining the hyperactive cellular proliferation characteristic of aggressive AML.
The implications of these findings are profound. Targeting SETD1B’s catalytic domain or its enzymatic activity presents a novel therapeutic strategy specifically tailored to combat AML subtypes refractory to current treatments. Existing compounds like Chaetocin, known to inhibit related methyltransferases, may serve as valuable starting points in designing selective SETD1B inhibitors. The development of such targeted epigenetic therapies could potentially disrupt the leukemia’s transcriptional addiction to MYC, thereby impairing the cancer’s ability to proliferate unchecked.
Moreover, as SETD1B activity correlates with leukemia aggressiveness, measuring its levels or functional state could emerge as an essential biomarker to guide treatment decisions. Stratifying patients based on SETD1B expression or H3K4me3 patterns could identify those most likely to benefit from epigenetic therapies, enabling a precision medicine approach in AML treatment. The study also opens the door to investigating SETD1B’s role in other MYC-driven cancers, broadening the scope of epigenetic therapies beyond hematological malignancies.
This innovative research not only furthers our understanding of how epigenetic modifications contribute to leukemogenesis but also highlights the intricate interplay between genetic mutations like FLT3-ITD and chromatin state in shaping cancer behavior. The exploitation of such epigenetic vulnerabilities holds promise for overcoming therapeutic resistance—a formidable challenge in AML management.
Dr. Hoshii and his colleagues emphasize the need for continued exploration into the molecular machinery that sustains oncogenic transcription. Their work exemplifies how basic science discoveries in chromatin biology can translate into actionable targets for drug development, offering new hope for patients facing the bleak prognosis of FLT3-ITD-mutated AML.
In conclusion, the study published in Leukemia delineates a critical epigenetic axis involving SETD1B-mediated H3K4me3 dynamics and MYC regulation as a driver of aggressive AML phenotypes. This breakthrough offers a compelling rationale for developing inhibitors against SETD1B’s catalytic activity and for incorporating epigenetic biomarkers in clinical oncology. As the scientific community continues to unravel the complexities of cancer epigenomes, such findings underscore the potential of epigenetic therapies to revolutionize cancer treatment paradigms.
Subject of Research: Cells
Article Title: Regulation of H3K4me3 breadth and MYC expression by the SETD1B catalytic domain in MLL-rearranged leukemia
News Publication Date: 8-May-2025
Web References:
DOI: 10.1038/s41375-025-02638-y
Image Credits: Dr. Takayuki Hoshii from Chiba University, Japan
Keywords: Acute myeloid leukemia, AML, FLT3-ITD mutation, SETD1B, epigenetics, H3K4me3, MYC oncogene, catalytic domain, chromatin modification, CRISPR screening, leukemia therapy, epigenetic therapy
Tags: acute myeloid leukemia researchadvancements in hematological malignancies.bone marrow malignancieschemotherapy resistance in acute myeloid leukemiaepigenetic mechanisms in cancerFLT3 internal tandem duplication mutationhistone modifications in AMLMYC proto-oncogene and cancerpatient outcomes in leukemia treatmentrelapse rates in blood cancerSETD1B role in leukemiatargeted treatments for aggressive blood cancer

{kind=link}