Credit: Chris Glembotski
A protein found in the heart that is known to be involved in cellular stress responses in cancer cells is now believed to play a critical role in the ability of cardiac cells to combat heart disease and recover from a heart attack. A new study led by San Diego State University molecular cardiologist Christopher Glembotski, director of the SDSU Heart Institute, found that the protein appears to promote the natural ability of heart cells to ward off stress-induced damage. This finding suggests a novel treatment and prevention strategy for people at risk of heart disease, according to Glembotski's research.
The protein, known as ATF6, occurs naturally in all cells in the human body. Research by Glembotski's group and others over the years has shown that ATF6 responds to stress brought on by misfolded proteins in a part of the cell called the endoplasmic reticulum. This stress is often caused by an overabundance of reactive oxygen molecules that derail the cell's normal ability to function–the primary reason for damage to the heart when people suffer from heart disease and heart attacks. In previous studies, Glembotski and his colleagues showed that during heart attacks in mice, ATF6 is called into action, but its function in the heart was not known.
To investigate this mystery, Glembotski and his colleagues turned to a strain of mice that lack the gene that codes for the production of ATF6. In these mice, they found that heart disease caused more extensive damage than mice with normally functioning versions of the gene.
Looking further into what other effects the ATF6 protein had in cells, the researchers discovered that it activated a cascade of stress-response genes not previously known to be associated with ATF6. These genes produced an enzyme known as catalase, which acts as an antioxidant and neutralizes harmful reactive oxygen molecules, reducing cellular stress and preventing proteins from misfolding in the first place. By artificially introducing catalase into mice that lacked the ATF6 gene, Glembotski and his team found that these treated mice showed the same protective effects as mice with a working version of the ATF6 gene.
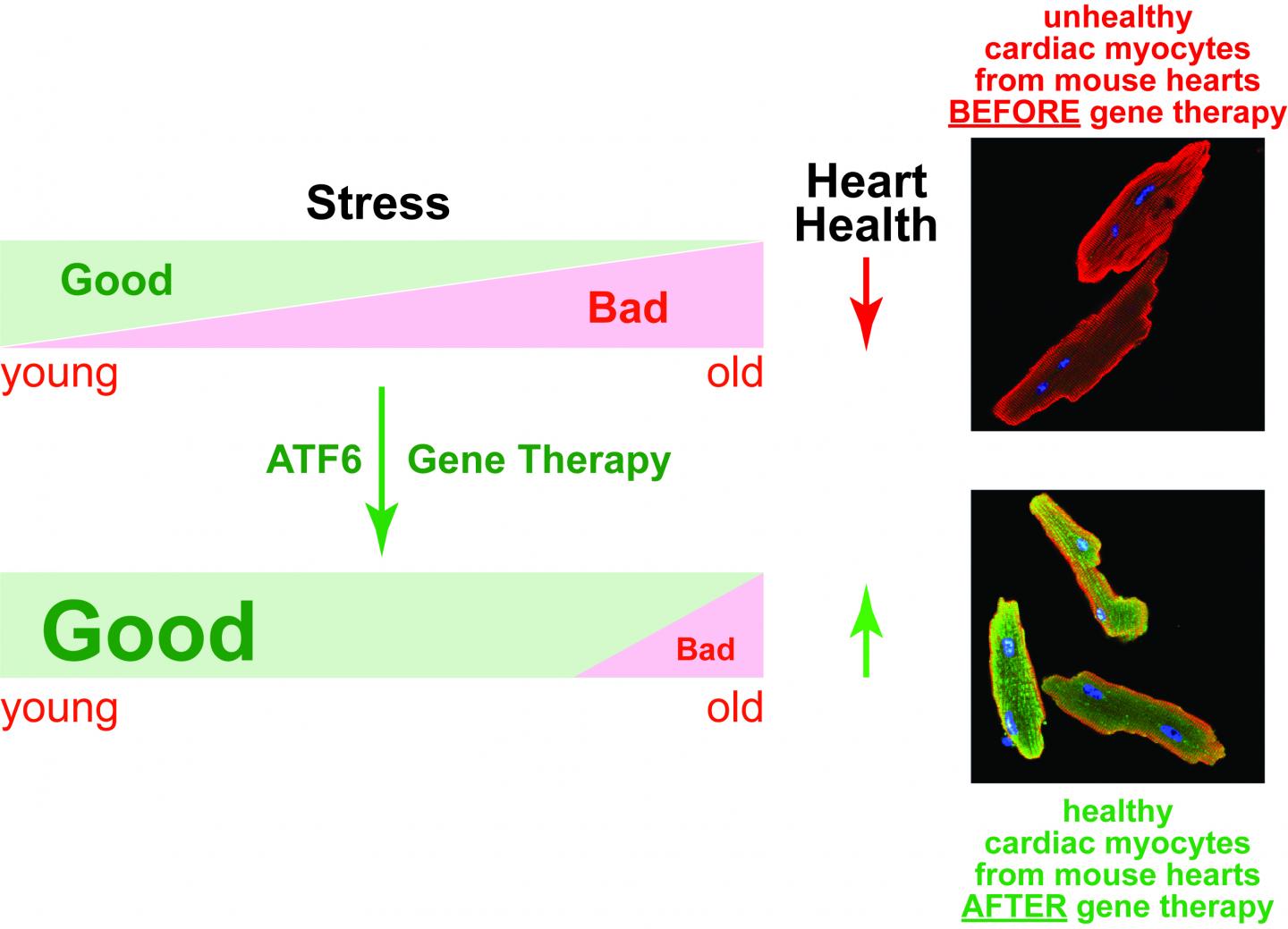
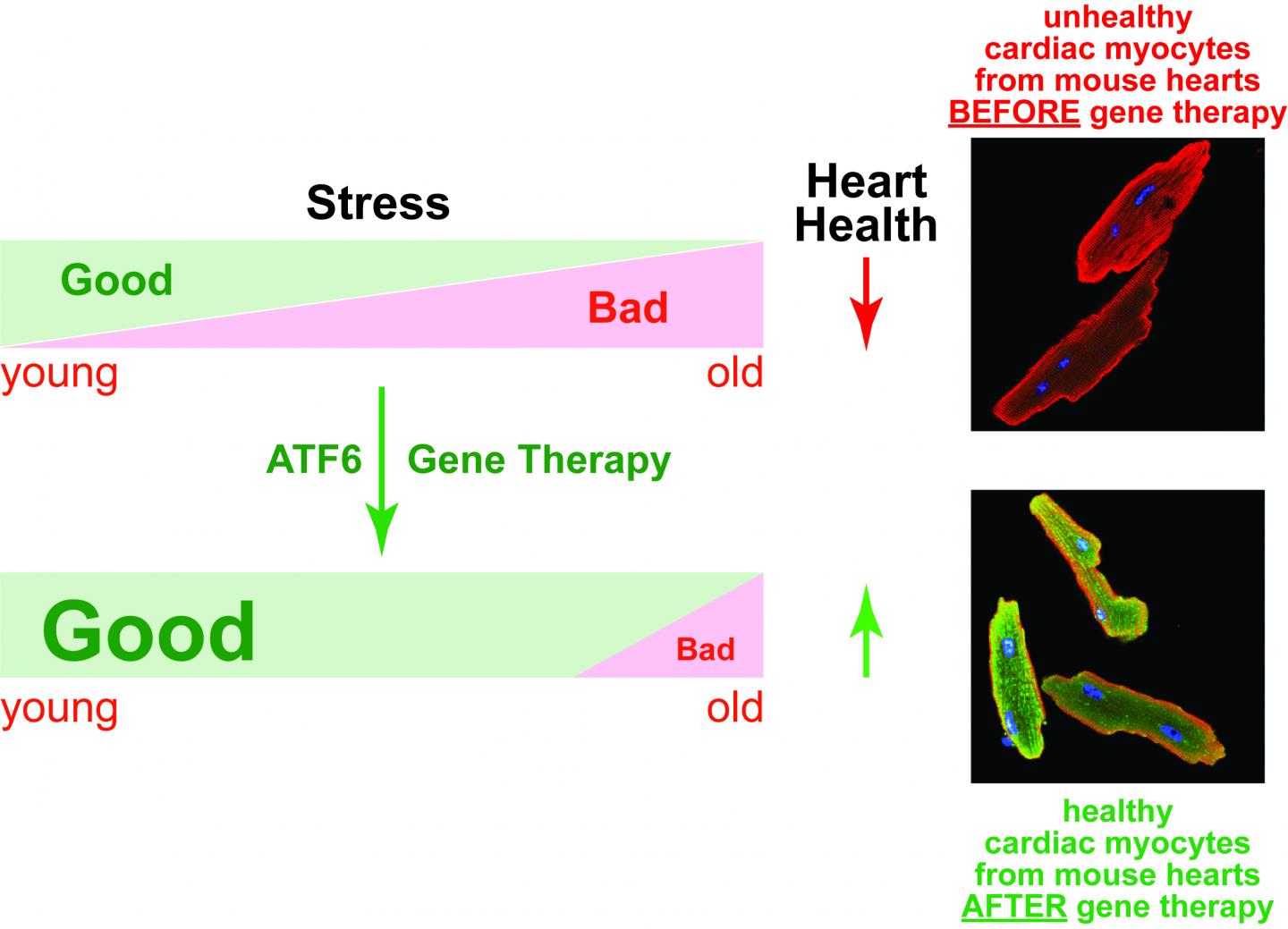
The team went on to show that as mice age, they progressively lose ATF6, and their hearts become more prone to damage during a heart attack. In an effort to arrest this age-related effect, Glembotski and his team developed a drug based on gene therapy that could boost ATF6 production in heart cells. It worked, and the older mice who received the drug showed less damage following a heart attack.
"These cellular mechanics should work very much the same way in humans," Glembotski said. Taken together, the results suggest that ATF6 plays a critical role in jumpstarting a process that reduces ischemia/reperfusion damage (I/R). I/R occurs when blood rushes back into tissue after a heart attack. The researchers reported their findings in the March 3 edition of the journal Circulation Research.
"It seems to be an adaptive response of the heart that is lost with age," Glembotski said. "We think that, like the mice, human heart cells usually make some ATF6, but if they could make more–like in the young heart–the heart would be more resistant to heart disease."
To that end, he and his lab–which has been funded continuously by the National Institutes of Health since 1983–are looking at various ways to boost ATF6 production in the heart. One simple way to do so may be increased exercising.
"Our preliminary studies show that exercise might naturally boost endogenous ATF6 and help prevent damage from I/R," Glembotski said.
An additional way of increasing ATF6 in the heart might involve using a gene therapy to boost naturally occurring ATF6. This could be especially effective for older people whose natural protection against cellular stress has waned.
"As we age, the adaptive stress response in the heart decreases," Glembotski said. "If we can deliver new versions of these genes to the heart, we could bring back some of that protection that you naturally have when you are young."
###
Media Contact
Mike Price
[email protected]
@SDSU
http://www.sdsu.edu
############
Story Source: Materials provided by Scienmag

{kind=link}