In an era where acute kidney injury (AKI) continues to represent a formidable clinical challenge with limited therapeutic options, groundbreaking research is shedding light on the molecular underpinnings of oxalate-induced renal damage. A recent study published in Cell Death Discovery by Ye, Lan, Chen, and colleagues pioneers a detailed exploration into the intricate roles of ACSL4, GPX4, and FSP1 enzymes within the context of oxalate toxicity, providing unprecedented insights into the pathophysiology of kidney injury and unveiling promising avenues for targeted intervention.
Acute kidney injury induced by oxalate crystals remains a significant contributor to renal morbidity worldwide, often leading to rapid deterioration of kidney function and, in severe cases, the need for renal replacement therapy. Despite recognition of oxalate accumulation as a pivotal injurious factor, the precise molecular cascades facilitating renal parenchymal damage have long remained elusive. The innovative work by Ye et al. pivots on unraveling how lipid metabolism and ferroptosis-related pathways converge to exacerbate oxalate-triggered AKI.
Central to this study are the enzymes ACSL4 (acyl-CoA synthetase long-chain family member 4), GPX4 (glutathione peroxidase 4), and FSP1 (ferroptosis suppressor protein 1). These proteins interact within the lipid peroxidation and ferroptosis framework, a regulated cell death modality increasingly implicated in diverse pathologies, especially those characterized by oxidative stress and inflammation. By focusing on these molecular players, the researchers delineated how oxidative lipid damage and ferroptosis drive tubular epithelial cell injury in response to oxalate overload.
.adsslot_S1GU43MQdr{ width:728px !important; height:90px !important; }
@media (max-width:1199px) { .adsslot_S1GU43MQdr{ width:468px !important; height:60px !important; } }
@media (max-width:767px) { .adsslot_S1GU43MQdr{ width:320px !important; height:50px !important; } }
ADVERTISEMENT
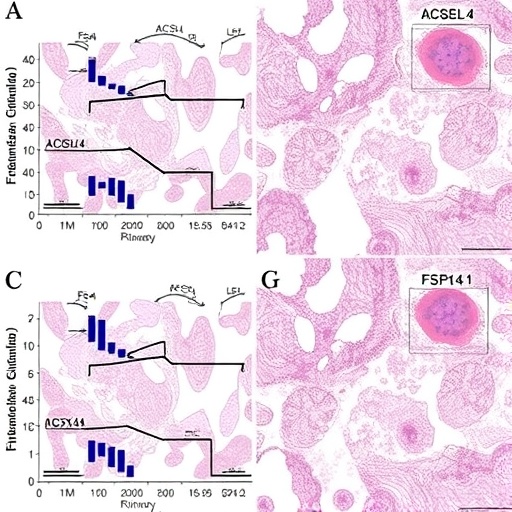
The team employed a comprehensive array of experimental approaches, including in vivo murine models of oxalate-induced AKI, alongside in vitro renal tubular cell cultures exposed to pathological oxalate concentrations. Through meticulous gene expression analyses, immunohistochemistry, and functional assays, the researchers documented a marked upregulation of ACSL4 concomitant with a downregulation of GPX4 and FSP1 activity during the acute phase of injury. This imbalance precipitates a surge in lipid peroxides, destabilizing cellular membranes and exacerbating cell death.
Intriguingly, ACSL4, traditionally known for catalyzing the activation of polyunsaturated fatty acids into acyl-CoA derivatives, emerged as a critical promoter of ferroptosis under oxalate stress. Elevated ACSL4 expression facilitates incorporation of oxidizable fatty acids into membrane phospholipids, sensitizing tubular epithelial cells to lipid peroxidation. Conversely, the antioxidant enzyme GPX4 exerts a protective effect by reducing lipid hydroperoxides, thereby staving off ferroptotic cascades. The observed depletion of GPX4, paired with diminished FSP1 levels, undermines cellular defenses, rendering the kidney parenchyma vulnerable to oxidative injury.
Beyond cell death, the interplay between these enzymes orchestrates inflammatory signaling and fibrotic remodeling in the damaged kidney. The enhanced oxidative milieu triggered by ACSL4-driven lipid peroxidation propagates pro-inflammatory cytokine secretion and immune cell recruitment, aggravating tissue injury. The protective axis mediated by GPX4 and FSP1, therefore, also serves to quell inflammation and preserve renal architecture, implicating ferroptosis regulators as key gatekeepers of the renal microenvironment.
The molecular definition of ferroptosis in kidney injury expands our understanding beyond classical apoptosis and necrosis, challenging existing paradigms and enriching the tapestry of regulated cell death modalities implicated in renal pathology. Traditionally, therapeutic interventions have largely aimed at symptom management and supportive care; however, the emerging characterization of ferroptosis effectors opens a new frontier for kidney-directed molecular therapies with specificity and efficacy.
Intriguingly, this research also dovetails with burgeoning evidence implicating ferroptosis across systemic diseases characterized by oxidative stress, including neurodegeneration, cardiovascular disorders, and cancer. The kidney, due to its high metabolic demands and exposure to circulating toxins such as oxalate, represents a critical organ in which ferroptosis findings may be particularly impactful and rapidly translatable into clinical benefits.
From a mechanistic perspective, the study meticulously interlinks ACSL4-mediated fatty acid metabolism alterations with the depletion of glutathione-dependent defenses governed by GPX4 and FSP1, creating a detailed molecular landscape of susceptibility and protection. This integrative view enriches the foundational knowledge of lipid homeostasis dysregulation in kidney disease and may inform the design of combinational therapies targeting multiple nodes within the ferroptosis network.
Furthermore, the authors emphasize the need to consider patient heterogeneity, noting that genetic polymorphisms in ACSL4, GPX4, or FSP1 might modulate individual susceptibility to oxalate nephropathy and response to ferroptosis-targeted therapies. Future research, therefore, must integrate genomic profiling with phenotypic characterization to refine personalized medicine approaches in AKI management.
At the translational level, this work galvanizes the development of novel biomarkers reflecting ferroptotic activity, which could revolutionize early diagnosis and prognostic assessment in oxalate-related renal injury. Measuring circulating or urinary lipid peroxidation products, or the expression profiles of ACSL4, GPX4, and FSP1, may yield sensitive indicators enabling timely therapeutic intervention before irreversible damage ensues.
In conclusion, the seminal findings of Ye et al. signify a paradigm shift in our comprehension of oxalate-induced AKI, illuminating ferroptosis as a central pathological mechanism while pinpointing ACSL4, GPX4, and FSP1 as master regulators. This knowledge paves the way for innovative, mechanism-based therapies targeting lipid peroxidation and ferroptotic cell death, ultimately aiming to transform patient outcomes in a domain historically bereft of effective treatments.
As the global burden of chronic kidney disease and episodes of acute kidney injury continues to rise, the translational implications of this research resonate far beyond the laboratory. By harnessing the nuanced molecular pathways unveiled in this study, the medical community can aspire toward tailored, robust interventions that not only halt disease progression but also restore renal health with precision.
This compelling advancement exemplifies the power of interdisciplinary science—blending cell biology, nephrology, and molecular pharmacology—to redefine disease paradigms and inspire a future where acute kidney injury becomes a manageable and reversible condition rather than a devastating clinical endpoint. The implications of this study will undoubtedly ripple through renal medicine, influencing research, clinical practice, and ultimately, the lives of patients worldwide.
Subject of Research: Roles of ACSL4, GPX4, and FSP1 in oxalate-induced acute kidney injury and ferroptosis mechanisms
Article Title: Roles of ACSL4/GPX4 and FSP1 in oxalate-induced acute kidney injury
Article References:
Ye, K., Lan, R., Chen, Z. et al. Roles of ACSL4/GPX4 and FSP1 in oxalate-induced acute kidney injury. Cell Death Discov. 11, 279 (2025). https://doi.org/10.1038/s41420-025-02557-y
Image Credits: AI Generated
DOI: https://doi.org/10.1038/s41420-025-02557-y
Tags: ACSL4 role in kidney injuryenzyme interactions in renal pathologyferroptosis and kidney diseaseFSP1 and renal healthGPX4 function in oxalate toxicitylipid metabolism in renal damagemechanisms of acute kidney injurymolecular pathways in kidney dysfunctionoxalate crystals and renal morbidityoxalate-induced renal injury researchoxidative stress in kidney injuriestherapeutic targets for AKI
{kind=link}