Researchers led by the University of Missouri create a new method for analyzing large amounts of biological data to help scientists draw faster conclusions for possible treatments.
Credit: University of Missouri
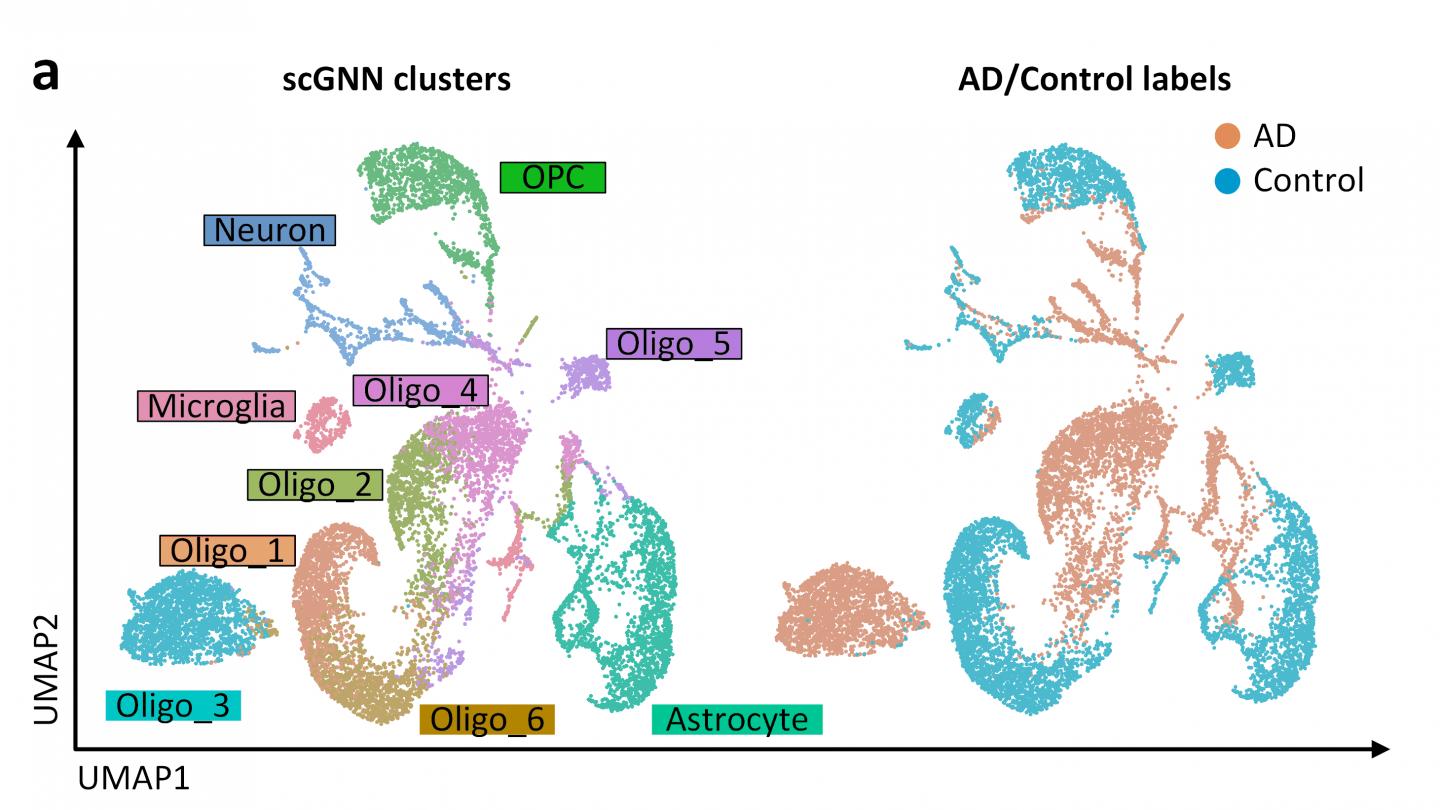
Studying genetic material on a cellular level, such as single-cell RNA-sequencing, can provide scientists with a detailed, high-resolution view of biological processes at work. This level of detail helps scientists determine the health of tissues and organs, and better understand the development of diseases such as Alzheimer’s that impacts millions of people. However, a lot of data is also generated, and leads to the need for an efficient, easy-to-use way to analyze it.
Now, a team of engineers and scientists from the University of Missouri and the Ohio State University have created a new way to analyze data from single-cell RNA-sequencing by using a computer method called “machine learning.” This method uses the power of computers to intelligently analyze large amounts of data and help scientists draw faster conclusions and move to the next stage of the research. Their methodology is detailed in a new paper published by Nature Communications.
“Single cell genetic profiling is on the cutting edge of today’s technological advances because it measures how many genes are present and how they are expressed from the level of an individual biological cell,” said Dong Xu, a professor in the MU College of Engineering. “At a minimum, there could be tens of thousands of cells being analyzed in this manner, so there ends up being a huge amount of data collected. Currently, determining conclusions from this type of data can be challenging because a lot of data must be filtered through in order to find what researchers are looking for. So, we applied one of the newest machine-learning methods to tackle this problem — a graph neural network.”
After computers intelligently analyze the data through a machine learning process, the graph neural network then takes the results and creates a visual representation of the data to help easily identify patterns. The graph is made up of dots — each dot representative of a cell — and similar types of cells are color coded for easy recognition. Xu said precision medicine is a good example of how single-cell RNA-sequencing can be used.
“With this data, scientists can study the interactions between cells within the micro-environment of a cancerous tissue, or watch the T-cells, B-cells and immune cells all try to attack the cancerous cells,” Xu said. “Therefore, in cases where a person has a strong immune system, and the cancer hasn’t fully developed yet, we can learn how the cancer can possibly be killed at an early stage, and we have our results sooner because of machine learning, which leads us to a viable treatment faster.”
Xu believes this is a great example of how engineers and biologists can work together to study problems or issues in biology. He hopes this method can be used by biologists as a new tool to help solve complex biological questions, such as a possible treatment for Alzheimer’s disease.
###
The study, “scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses,” was published in Nature Communications. Funding was provided by awards (R35-GM126985 and R01-GM131399) from the National Institute of General Medical Sciences of the National Institutes of Health.
Editor’s Note: Xu has joint appointments in the Electrical Engineering and Computer Science Department, the Christopher S. Bond Life Sciences Center and the Informatics Institute at the University of Missouri. Juexin Wang, a research scientist at MU, is also the first author on the study. Collaborators at the Ohio State University include Qin Ma, Anjun Ma and Hongjun Fu.
Media Contact
Eric Stann
[email protected]
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}